NAD+ 代謝及其在衰老過程中在細胞過程中的作用 (2021/02)
摘要
煙酰胺腺嘌呤二核苷酸 (NAD+) 是一種用於氧化還原反應的輔酶,是能量代謝的核心。 NAD+ 也是非氧化還原 NAD+ 依賴性酶的重要輔助因子,包括去乙酰化酶、CD38 和聚(ADP-核糖)聚合酶。 NAD+ 可以直接和間接影響許多關鍵的細胞功能,包括代謝途徑、DNA 修復、染色質重塑、細胞衰老和免疫細胞功能。這些細胞過程和功能對於維持組織和代謝穩態以及健康老齡化至關重要。值得注意的是,在包括囓齒動物和人類在內的多種模式生物中,衰老伴隨著組織和細胞 NAD+ 水平的逐漸下降。 NAD+ 水平的下降與許多與衰老相關的疾病有因果關係,包括認知能力下降、癌症、代謝疾病、肌肉減少症和虛弱。許多這些與衰老相關的疾病可以通過恢復 NAD+ 水平來減緩甚至逆轉。因此,靶向 NAD+ 代謝已成為改善衰老相關疾病、延長人類健康壽命和壽命的潛在治療方法。然而,關於 NAD+ 如何影響人類健康和衰老生物學還有很多需要了解。這包括更深入地了解調節 NAD+ 水平的分子機制、如何在衰老過程中有效恢復 NAD+ 水平、這樣做是否安全以及補充 NAD+ 是否會對人類衰老產生有益影響。
煙酰胺腺嘌呤二核苷酸 (NAD+) 是氧化還原反應的重要輔酶,是能量代謝的核心。 NAD+ 也是非氧化還原 NAD+ 依賴性酶(包括去乙酰化酶和聚(ADP-核糖)聚合酶 (PARP))的重要輔助因子。
NAD+ 最初因其在調節酵母提取物中代謝率中的作用而被確定,後來成為氧化還原反應中的主要氫化物受體。 NAD+ 接受氫陰離子並形成其還原形式 NADH 的這種能力對於所有生命形式的代謝反應至關重要,並調節參與多種分解代謝途徑的脫氫酶的活性,包括糖酵解、谷氨酰胺分解和脂肪酸氧化。然後將這些反應中接受的電子捐贈給電子傳遞鏈,在真核生物中形成 ATP。 NAD+ 也可以被磷酸化形成 NADP+,NADP+ 也可作為氫化物受體形成 NADPH,並用於保護免受氧化應激和需要還原能力的合成代謝途徑,例如脂肪酸合成。
除了能量代謝之外,NAD+ 還被數百種酶用作輔助因子或底物,因此在調節細胞過程和細胞功能方面具有多種作用,其中許多仍在研究中。 NAD+ 水平與健康之間的聯繫是在近一個世紀前建立的。 1937 年,Conrad Elvehjem 發現糙皮病(以皮炎、腹瀉和癡呆為特徵)是由飲食中缺乏菸酸引起的,導致 NAD+ 和 NADP+ 水平低。最近,低 NAD+ 水平與多種疾病狀態有關,包括代謝和神經退行性疾病,現在已知較低的 NAD+ 水平與囓齒動物和人類的衰老有關。因此,人們對了解 NAD+ 代謝如何影響疾病的起源,特別是與衰老相關的疾病產生了新的興趣。在這方面,用 NAD+ 前體煙酰胺核苷 (NR) 和煙酰胺單核苷酸 (NMN) 恢復 NAD+ 水平已成為治療年齡相關疾病的重要治療方法,並且似乎在體內具有有益效果,至少在囓齒動物模型中是這樣。
在本次回顧中,我們將重點關注過去 5 年在 NAD+ 領域的工作。特別是,我們研究了 NAD+ 前體和最終 NAD+ 水平影響衰老和疾病狀態下的生理學和健康壽命的分子機制。這些機制可能是複雜的和多因素的,因此我們分多個部分討論它們。首先,我們回顧了關於如何在衰老過程中調節主要 NAD+ 生物合成和降解途徑的新發現。其次,我們討論了較低的 NAD+ 水平對與衰老相關的疾病很重要的分子過程的可能後果,包括 DNA 修復、表觀遺傳學和基因表達的調節、細胞代謝和氧化還原平衡的調節。第三,我們描述了衰老過程中 NAD+ 依賴的機制,包括代謝紊亂、免疫系統失調、細胞衰老和神經退行性變。最後,我們回顧了許多近期的高質量臨床前研究,這些研究調查了恢復 NAD+ 水平以治療衰老相關疾病的方法。其中包括大量使用 NAD+ 前體的研究,以及促進 NAD+ 生物合成的小分子藥物。最後,我們討論了這些不同的策略和這些研究的結果,以概述基於調節 NAD+ 水平的療法在促進人類健康和壽命方面的前景。
1. 細胞 NAD+ 代謝
NAD+ 在細胞質、線粒體和細胞核中高度劃分,代表其主要的亞細胞池(補充框 1)。這些池相互獨立調節,與此一致的是,參與 NAD+ 生物合成或降解的酶也高度分區 。 NAD+ 是多種代謝途徑和細胞過程的關鍵代謝物和輔酶。首先,NAD+ 的減少是維持細胞能量平衡和氧化還原狀態所必需的。 NAD+ 還被三類 NAD+ 消耗酶不斷轉化:NAD+ 糖水解酶,也稱為 NADases(CD38、CD157 和 SARM1),sirtuins 和 PARP 的蛋白質脫酰基酶家族,具有各種重要的細胞功能。它們利用 NAD+ 作為底物或輔因子,並產生煙酰胺 (NAM) 作為副產物(圖 1)。因此,NAD+ 介導了多個主要的生物過程,並且總是需求量很大(補充框 2 和 3)。為了維持 NAD+ 水平,NAM 可以通過 NAM 回收途徑回收回 NAD+(詳情參見框 1)。此外,一些細胞,主要是在肝臟中,可以從多種飲食來源從頭合成 NAD+。因此,NAD+ 在細胞中不斷合成、分解代謝和再循環,以維持穩定的細胞內 NAD+ 水平(圖 1)。然而,在衰老過程中,分解代謝和合成代謝過程之間的這種平衡可能會發生變化,NAD+ 降解可能超過細胞從頭產生 NAD+ 的能力或其有效回收或挽救 NAM 的能力。此外,過量的 NAM 可能通過替代代謝途徑分解代謝(詳情參見框 2),有效地將其從 NAM 補救途徑轉移並進一步影響 NAD+ 水平。除了作為 NAD+ 消耗酶的共同作用外,NAD+ 糖水解酶、sirtuins 和 PARP 在衰老和與年齡相關的疾病中具有不同的作用。雖然增強 sirtuins 的激活已成為延長壽命和健康壽命的一種方式,但 PARP 和 NAD+ 糖水解酶(如 CD38)的異常激活可能會產生相反的效果並加劇衰老表型(見下文)。
煙酰胺腺嘌呤二核苷酸 (NAD+) 是一種用於氧化還原反應的輔酶,是能量代謝的核心。 NAD+ 也是非氧化還原 NAD+ 依賴性酶的重要輔助因子,包括去乙酰化酶、CD38 和聚(ADP-核糖)聚合酶。 NAD+ 可以直接和間接影響許多關鍵的細胞功能,包括代謝途徑、DNA 修復、染色質重塑、細胞衰老和免疫細胞功能。這些細胞過程和功能對於維持組織和代謝穩態以及健康老齡化至關重要。值得注意的是,在包括囓齒動物和人類在內的多種模式生物中,衰老伴隨著組織和細胞 NAD+ 水平的逐漸下降。 NAD+ 水平的下降與許多與衰老相關的疾病有因果關係,包括認知能力下降、癌症、代謝疾病、肌肉減少症和虛弱。許多這些與衰老相關的疾病可以通過恢復 NAD+ 水平來減緩甚至逆轉。因此,靶向 NAD+ 代謝已成為改善衰老相關疾病、延長人類健康壽命和壽命的潛在治療方法。然而,關於 NAD+ 如何影響人類健康和衰老生物學還有很多需要了解。這包括更深入地了解調節 NAD+ 水平的分子機制、如何在衰老過程中有效恢復 NAD+ 水平、這樣做是否安全以及補充 NAD+ 是否會對人類衰老產生有益影響。
煙酰胺腺嘌呤二核苷酸 (NAD+) 是氧化還原反應的重要輔酶,是能量代謝的核心。 NAD+ 也是非氧化還原 NAD+ 依賴性酶(包括去乙酰化酶和聚(ADP-核糖)聚合酶 (PARP))的重要輔助因子。
NAD+ 最初因其在調節酵母提取物中代謝率中的作用而被確定,後來成為氧化還原反應中的主要氫化物受體。 NAD+ 接受氫陰離子並形成其還原形式 NADH 的這種能力對於所有生命形式的代謝反應至關重要,並調節參與多種分解代謝途徑的脫氫酶的活性,包括糖酵解、谷氨酰胺分解和脂肪酸氧化。然後將這些反應中接受的電子捐贈給電子傳遞鏈,在真核生物中形成 ATP。 NAD+ 也可以被磷酸化形成 NADP+,NADP+ 也可作為氫化物受體形成 NADPH,並用於保護免受氧化應激和需要還原能力的合成代謝途徑,例如脂肪酸合成。
除了能量代謝之外,NAD+ 還被數百種酶用作輔助因子或底物,因此在調節細胞過程和細胞功能方面具有多種作用,其中許多仍在研究中。 NAD+ 水平與健康之間的聯繫是在近一個世紀前建立的。 1937 年,Conrad Elvehjem 發現糙皮病(以皮炎、腹瀉和癡呆為特徵)是由飲食中缺乏菸酸引起的,導致 NAD+ 和 NADP+ 水平低。最近,低 NAD+ 水平與多種疾病狀態有關,包括代謝和神經退行性疾病,現在已知較低的 NAD+ 水平與囓齒動物和人類的衰老有關。因此,人們對了解 NAD+ 代謝如何影響疾病的起源,特別是與衰老相關的疾病產生了新的興趣。在這方面,用 NAD+ 前體煙酰胺核苷 (NR) 和煙酰胺單核苷酸 (NMN) 恢復 NAD+ 水平已成為治療年齡相關疾病的重要治療方法,並且似乎在體內具有有益效果,至少在囓齒動物模型中是這樣。
在本次回顧中,我們將重點關注過去 5 年在 NAD+ 領域的工作。特別是,我們研究了 NAD+ 前體和最終 NAD+ 水平影響衰老和疾病狀態下的生理學和健康壽命的分子機制。這些機制可能是複雜的和多因素的,因此我們分多個部分討論它們。首先,我們回顧了關於如何在衰老過程中調節主要 NAD+ 生物合成和降解途徑的新發現。其次,我們討論了較低的 NAD+ 水平對與衰老相關的疾病很重要的分子過程的可能後果,包括 DNA 修復、表觀遺傳學和基因表達的調節、細胞代謝和氧化還原平衡的調節。第三,我們描述了衰老過程中 NAD+ 依賴的機制,包括代謝紊亂、免疫系統失調、細胞衰老和神經退行性變。最後,我們回顧了許多近期的高質量臨床前研究,這些研究調查了恢復 NAD+ 水平以治療衰老相關疾病的方法。其中包括大量使用 NAD+ 前體的研究,以及促進 NAD+ 生物合成的小分子藥物。最後,我們討論了這些不同的策略和這些研究的結果,以概述基於調節 NAD+ 水平的療法在促進人類健康和壽命方面的前景。
1. 細胞 NAD+ 代謝
NAD+ 在細胞質、線粒體和細胞核中高度劃分,代表其主要的亞細胞池(補充框 1)。這些池相互獨立調節,與此一致的是,參與 NAD+ 生物合成或降解的酶也高度分區 。 NAD+ 是多種代謝途徑和細胞過程的關鍵代謝物和輔酶。首先,NAD+ 的減少是維持細胞能量平衡和氧化還原狀態所必需的。 NAD+ 還被三類 NAD+ 消耗酶不斷轉化:NAD+ 糖水解酶,也稱為 NADases(CD38、CD157 和 SARM1),sirtuins 和 PARP 的蛋白質脫酰基酶家族,具有各種重要的細胞功能。它們利用 NAD+ 作為底物或輔因子,並產生煙酰胺 (NAM) 作為副產物(圖 1)。因此,NAD+ 介導了多個主要的生物過程,並且總是需求量很大(補充框 2 和 3)。為了維持 NAD+ 水平,NAM 可以通過 NAM 回收途徑回收回 NAD+(詳情參見框 1)。此外,一些細胞,主要是在肝臟中,可以從多種飲食來源從頭合成 NAD+。因此,NAD+ 在細胞中不斷合成、分解代謝和再循環,以維持穩定的細胞內 NAD+ 水平(圖 1)。然而,在衰老過程中,分解代謝和合成代謝過程之間的這種平衡可能會發生變化,NAD+ 降解可能超過細胞從頭產生 NAD+ 的能力或其有效回收或挽救 NAM 的能力。此外,過量的 NAM 可能通過替代代謝途徑分解代謝(詳情參見框 2),有效地將其從 NAM 補救途徑轉移並進一步影響 NAD+ 水平。除了作為 NAD+ 消耗酶的共同作用外,NAD+ 糖水解酶、sirtuins 和 PARP 在衰老和與年齡相關的疾病中具有不同的作用。雖然增強 sirtuins 的激活已成為延長壽命和健康壽命的一種方式,但 PARP 和 NAD+ 糖水解酶(如 CD38)的異常激活可能會產生相反的效果並加劇衰老表型(見下文)。
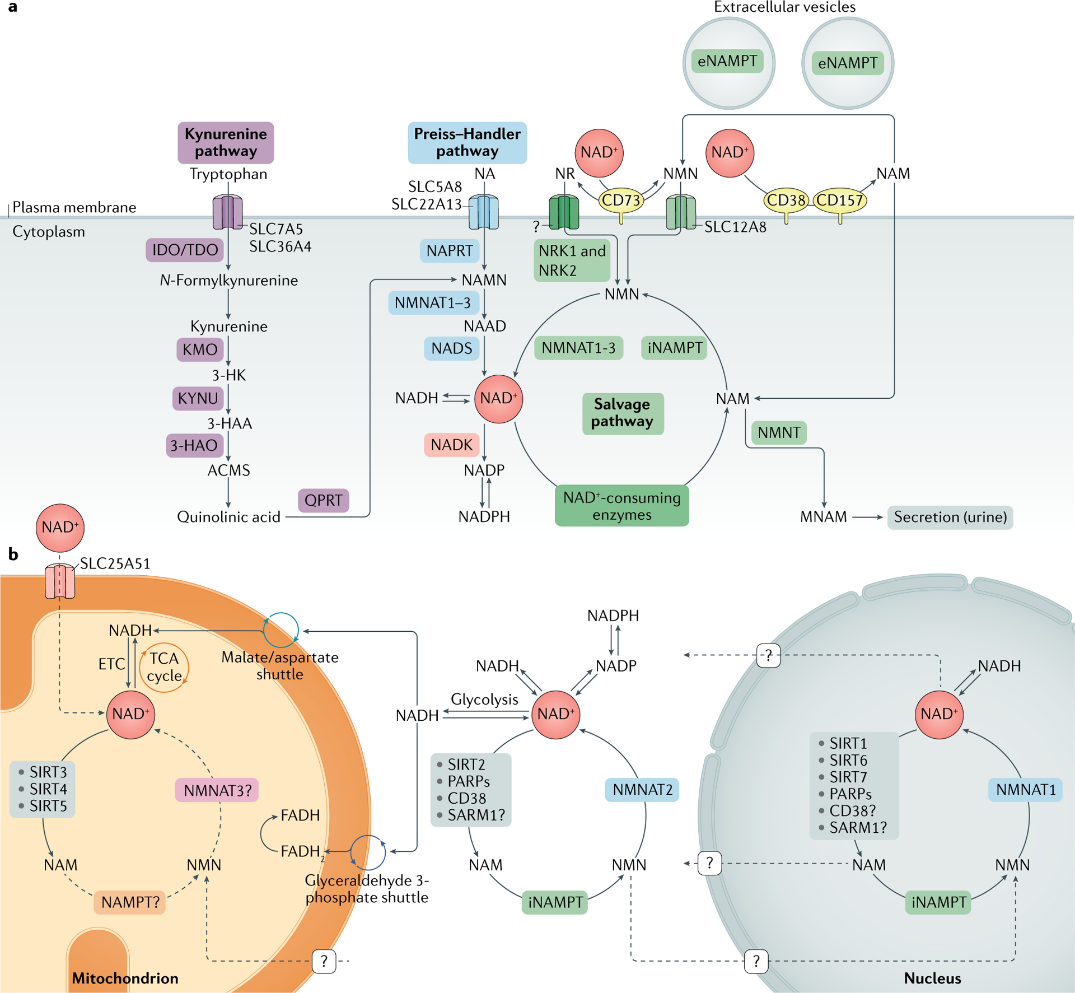
圖一 NAD+ 代謝。
A.一個煙酰胺腺嘌呤二核苷酸 (NAD+) 生物合成途徑。
NAD+ 水平由三個獨立的生物合成途徑維持。犬尿氨酸途徑(或從頭合成途徑)使用膳食氨基酸色氨酸生成 NAD+。色氨酸通過轉運蛋白 SLC7A5 和 SLC36A4 進入細胞。在細胞內,色氨酸通過限速酶吲哚胺 2,3-雙加氧酶 (IDO) 或限速酶色氨酸 2,3-雙加氧酶 (TDO) 轉化為 N-甲酰基犬尿氨酸。 N-甲酰基犬尿氨酸轉化為 L-犬尿氨酸,L-犬尿氨酸通過犬尿氨酸 3-單加氧酶 (KMO) 進一步轉化為 3-羥基犬尿氨酸 (3-HK),並通過色氨酸 2,3-雙加氧酶進一步轉化為 3-羥基鄰氨基苯甲酸 (3-HAA)。崑崙)。下一步由 3-羥基鄰氨基苯甲酸加氧酶 (3HAO) 生成 α-氨基-β-羧基粘康酸 ε-半醛 (ACMS)。該化合物可以自發縮合併重排為喹啉酸,其被喹啉酸磷酸核糖基轉移酶 (QPRT) 轉化為煙酰胺單核苷酸 (NAMN),此時它與 Preiss-Handler 途徑會聚。
Preiss-Handler 途徑使用膳食菸酸 (NA),通過 SLC5A8 或 SLC22A13 轉運蛋白進入細胞,以及酶菸酸磷酸核糖基轉移酶 (NAPRT) 生成 NAMN,然後通過煙酰胺轉化為菸酸腺嘌呤二核苷酸 (NAAD)單核苷酸腺苷酸轉移酶(NMNAT1、NMNAT2 和 NMNAT3)。該過程通過 NAD+ 合成酶 (NADS) 將 NAAD 轉化為 NAD+ 來完成。
NAD+ 補救途徑回收作為 NAD+ 消耗酶(sirtuins、聚(ADP-核糖)聚合酶 (PARPs) 和 NAD+ 糖水解酶和環狀 ADP-核糖合酶 CD38 的酶活性的副產物產生的煙酰胺 (NAM), CD157 和 SARM1)。最初,細胞內煙酰胺磷酸核糖基轉移酶 (iNAMPT) 將 NAM 再循環為煙酰胺單核苷酸 (NMN),然後通過不同的 NMNAT 將其轉化為 NAD+。 NAM 也可以被煙酰胺 N-甲基轉移酶 (NNMT) 甲基化並通過尿液分泌。在細胞外空間中,NAM 作為胞外酶 CD38 和 CD157 的副產物產生,並且可以通過細胞外 NAMPT (eNAMPT) 轉化為 NMN。然後 NMN 被 CD73 去磷酸化為煙酰胺核苷 (NR),然後通過未知的核苷轉運蛋白(問號)將其轉運到細胞中。 NMN 可以通過 NMN 特異性轉運蛋白(小腸中的 SLC12A8)導入細胞。在細胞內,NR 通過煙酰胺核苷激酶 1 和 2(NRK1 和 NRK2)形成 NMN。 NMN 然後由 NMNAT1、NMNAT2 和 NMNAT3 轉換為 NAD+。
B.不同亞細胞區室中的 NAD+ 代謝。
NAD+ 穩態是不同亞細胞區室中合成、消耗和再生的平衡,受亞細胞特異性 NAD+ 消耗酶、亞細胞轉運蛋白和氧化還原反應的調節。 NAD+ 前體通過三種生物合成途徑進入細胞(a 部分)。在細胞質中,NAM 通過細胞內 NAMPT (iNAMPT) 轉化為 NMN。然後 NMN 被 NMNAT2 轉化為 NAD+,NMNAT2 是這種酶的細胞質特異性異構體。 NAD+ 在糖酵解過程中被利用,產生 NADH,NADH 通過蘋果酸/天冬氨酸穿梭和甘油醛 3-磷酸穿梭轉移到線粒體基質。通過蘋果酸/天冬氨酸穿梭輸入的線粒體 NADH 被電子傳遞鏈 (ETC) 中的複合物 I 氧化,而來自甘油醛 3-磷酸穿梭的還原黃素腺嘌呤二核苷酸 (FADH2) 被複合物 II 氧化。最近鑑定了哺乳動物 NAD+ 線粒體轉運蛋白 SLC25A51,並已證明它負責細胞器中完整的 NAD+ 攝取。線粒體中 NAD+ 的補救途徑尚未完全解決,但已經提出了特定 NMNAT 異構體 (NMNAT3) 的作用。在線粒體中,NAD+ 被 NAD+ 依賴性線粒體 SIRT3–SIRT5 消耗,產生 NAM。目前尚不清楚 NAM 是否可以轉化回 NMN 或可以在線粒體內轉化為 NAD+,或者其他前體是否可以通過線粒體膜運輸以促進 NAD+ 的合成。核NAD+池可能通過擴散通過核孔與胞質池平衡。然而,完整的動態在很大程度上仍未被探索。已經描述了一種核特異性 NMNAT 異構體 (NMNAT1),它是核 NAM 挽救 NAD+ 途徑的一部分。 MNAM,N1-甲基煙酰胺; TCA,三羧酸。
A.一個煙酰胺腺嘌呤二核苷酸 (NAD+) 生物合成途徑。
NAD+ 水平由三個獨立的生物合成途徑維持。犬尿氨酸途徑(或從頭合成途徑)使用膳食氨基酸色氨酸生成 NAD+。色氨酸通過轉運蛋白 SLC7A5 和 SLC36A4 進入細胞。在細胞內,色氨酸通過限速酶吲哚胺 2,3-雙加氧酶 (IDO) 或限速酶色氨酸 2,3-雙加氧酶 (TDO) 轉化為 N-甲酰基犬尿氨酸。 N-甲酰基犬尿氨酸轉化為 L-犬尿氨酸,L-犬尿氨酸通過犬尿氨酸 3-單加氧酶 (KMO) 進一步轉化為 3-羥基犬尿氨酸 (3-HK),並通過色氨酸 2,3-雙加氧酶進一步轉化為 3-羥基鄰氨基苯甲酸 (3-HAA)。崑崙)。下一步由 3-羥基鄰氨基苯甲酸加氧酶 (3HAO) 生成 α-氨基-β-羧基粘康酸 ε-半醛 (ACMS)。該化合物可以自發縮合併重排為喹啉酸,其被喹啉酸磷酸核糖基轉移酶 (QPRT) 轉化為煙酰胺單核苷酸 (NAMN),此時它與 Preiss-Handler 途徑會聚。
Preiss-Handler 途徑使用膳食菸酸 (NA),通過 SLC5A8 或 SLC22A13 轉運蛋白進入細胞,以及酶菸酸磷酸核糖基轉移酶 (NAPRT) 生成 NAMN,然後通過煙酰胺轉化為菸酸腺嘌呤二核苷酸 (NAAD)單核苷酸腺苷酸轉移酶(NMNAT1、NMNAT2 和 NMNAT3)。該過程通過 NAD+ 合成酶 (NADS) 將 NAAD 轉化為 NAD+ 來完成。
NAD+ 補救途徑回收作為 NAD+ 消耗酶(sirtuins、聚(ADP-核糖)聚合酶 (PARPs) 和 NAD+ 糖水解酶和環狀 ADP-核糖合酶 CD38 的酶活性的副產物產生的煙酰胺 (NAM), CD157 和 SARM1)。最初,細胞內煙酰胺磷酸核糖基轉移酶 (iNAMPT) 將 NAM 再循環為煙酰胺單核苷酸 (NMN),然後通過不同的 NMNAT 將其轉化為 NAD+。 NAM 也可以被煙酰胺 N-甲基轉移酶 (NNMT) 甲基化並通過尿液分泌。在細胞外空間中,NAM 作為胞外酶 CD38 和 CD157 的副產物產生,並且可以通過細胞外 NAMPT (eNAMPT) 轉化為 NMN。然後 NMN 被 CD73 去磷酸化為煙酰胺核苷 (NR),然後通過未知的核苷轉運蛋白(問號)將其轉運到細胞中。 NMN 可以通過 NMN 特異性轉運蛋白(小腸中的 SLC12A8)導入細胞。在細胞內,NR 通過煙酰胺核苷激酶 1 和 2(NRK1 和 NRK2)形成 NMN。 NMN 然後由 NMNAT1、NMNAT2 和 NMNAT3 轉換為 NAD+。
B.不同亞細胞區室中的 NAD+ 代謝。
NAD+ 穩態是不同亞細胞區室中合成、消耗和再生的平衡,受亞細胞特異性 NAD+ 消耗酶、亞細胞轉運蛋白和氧化還原反應的調節。 NAD+ 前體通過三種生物合成途徑進入細胞(a 部分)。在細胞質中,NAM 通過細胞內 NAMPT (iNAMPT) 轉化為 NMN。然後 NMN 被 NMNAT2 轉化為 NAD+,NMNAT2 是這種酶的細胞質特異性異構體。 NAD+ 在糖酵解過程中被利用,產生 NADH,NADH 通過蘋果酸/天冬氨酸穿梭和甘油醛 3-磷酸穿梭轉移到線粒體基質。通過蘋果酸/天冬氨酸穿梭輸入的線粒體 NADH 被電子傳遞鏈 (ETC) 中的複合物 I 氧化,而來自甘油醛 3-磷酸穿梭的還原黃素腺嘌呤二核苷酸 (FADH2) 被複合物 II 氧化。最近鑑定了哺乳動物 NAD+ 線粒體轉運蛋白 SLC25A51,並已證明它負責細胞器中完整的 NAD+ 攝取。線粒體中 NAD+ 的補救途徑尚未完全解決,但已經提出了特定 NMNAT 異構體 (NMNAT3) 的作用。在線粒體中,NAD+ 被 NAD+ 依賴性線粒體 SIRT3–SIRT5 消耗,產生 NAM。目前尚不清楚 NAM 是否可以轉化回 NMN 或可以在線粒體內轉化為 NAD+,或者其他前體是否可以通過線粒體膜運輸以促進 NAD+ 的合成。核NAD+池可能通過擴散通過核孔與胞質池平衡。然而,完整的動態在很大程度上仍未被探索。已經描述了一種核特異性 NMNAT 異構體 (NMNAT1),它是核 NAM 挽救 NAD+ 途徑的一部分。 MNAM,N1-甲基煙酰胺; TCA,三羧酸。
2. NAD+消耗路線
由sirtuins消費
自發現以來,sirtuins 受到了廣泛關注,因為它們調節關鍵的代謝過程、壓力反應和衰老生物學。哺乳動物 sirtuin 家族由七種基因和蛋白質 (SIRT1–SIRT7) 組成,具有不同的亞細胞定位(SIRT1 和 SIRT6 的細胞核;SIRT7 的核仁;SIRT3、SIRT4 和 SIRT5 的線粒體;SIRT1、SIRT2 和 SIRT5 的細胞質)(圖. 1b)、酶活性和下游目標。這些 NAD+ 依賴性酶的亞細胞定位強調了細胞內 NAD+ 池的局部波動(它們本身由 sirtuins 調節)可能選擇性地影響細胞器特異性 sirtuin 功能和細胞代謝。
Sirtuins 在我們的細胞中持續活躍。 SIRT1 和 SIRT2 似乎占基礎條件下 NAD+ 總消耗量的約三分之一。此外,NAD+ 水平的升高與禁食和熱量限制期間的 sirtuin 激活密切相關 。還值得注意的是,sirtuin 活性與生物鐘耦合,由此 SIRT1 和 SIRT6 調節核心時鐘轉錄因子和下游晝夜轉錄組的活性。此外,SIRT1 和 NAD+ 補救途徑的關鍵酶煙酰胺磷酸核糖基轉移酶 (NAMPT) (BOX 1) 是 NAD+ 水平晝夜節律調節的關鍵參與者,而 NAMPT 由提供反饋迴路的生物鐘調節,導致 NAD+ 水平的晝夜振盪。
酵母的早期研究證明了沉默調節蛋白在基因沉默中的作用, 這在 2000 年歸因於它們作為 NAD+ 依賴性脫乙酰酶的活性 。據報導,Sirtuin 酶促活性主要包括通過兩步過程從靶蛋白的賴氨酸殘基中去除乙酰基:首先 NAD+ 被裂解為 NAM 和 ADP-核糖,其次,靶蛋白上的乙酰基被轉移ADP-核糖,從而形成中間肽基-ADP-核糖。隨後釋放乙酰-ADP-核糖(圖2a)。此外,sirtuin 家族的一些成員介導非乙酰賴氨酸酰化(例如,琥珀酰化、丙二酰化和脂肪酸酰化),其細胞功能迄今知之甚少。 SIRT4 和 SIRT6 也作為 ADP-核糖基轉移酶發揮作用,這表明去乙酰化酶也通過蛋白質乙酰化調節以外的機制促進細胞調節,這需要進一步研究。
由sirtuins消費
自發現以來,sirtuins 受到了廣泛關注,因為它們調節關鍵的代謝過程、壓力反應和衰老生物學。哺乳動物 sirtuin 家族由七種基因和蛋白質 (SIRT1–SIRT7) 組成,具有不同的亞細胞定位(SIRT1 和 SIRT6 的細胞核;SIRT7 的核仁;SIRT3、SIRT4 和 SIRT5 的線粒體;SIRT1、SIRT2 和 SIRT5 的細胞質)(圖. 1b)、酶活性和下游目標。這些 NAD+ 依賴性酶的亞細胞定位強調了細胞內 NAD+ 池的局部波動(它們本身由 sirtuins 調節)可能選擇性地影響細胞器特異性 sirtuin 功能和細胞代謝。
Sirtuins 在我們的細胞中持續活躍。 SIRT1 和 SIRT2 似乎占基礎條件下 NAD+ 總消耗量的約三分之一。此外,NAD+ 水平的升高與禁食和熱量限制期間的 sirtuin 激活密切相關 。還值得注意的是,sirtuin 活性與生物鐘耦合,由此 SIRT1 和 SIRT6 調節核心時鐘轉錄因子和下游晝夜轉錄組的活性。此外,SIRT1 和 NAD+ 補救途徑的關鍵酶煙酰胺磷酸核糖基轉移酶 (NAMPT) (BOX 1) 是 NAD+ 水平晝夜節律調節的關鍵參與者,而 NAMPT 由提供反饋迴路的生物鐘調節,導致 NAD+ 水平的晝夜振盪。
酵母的早期研究證明了沉默調節蛋白在基因沉默中的作用, 這在 2000 年歸因於它們作為 NAD+ 依賴性脫乙酰酶的活性 。據報導,Sirtuin 酶促活性主要包括通過兩步過程從靶蛋白的賴氨酸殘基中去除乙酰基:首先 NAD+ 被裂解為 NAM 和 ADP-核糖,其次,靶蛋白上的乙酰基被轉移ADP-核糖,從而形成中間肽基-ADP-核糖。隨後釋放乙酰-ADP-核糖(圖2a)。此外,sirtuin 家族的一些成員介導非乙酰賴氨酸酰化(例如,琥珀酰化、丙二酰化和脂肪酸酰化),其細胞功能迄今知之甚少。 SIRT4 和 SIRT6 也作為 ADP-核糖基轉移酶發揮作用,這表明去乙酰化酶也通過蛋白質乙酰化調節以外的機制促進細胞調節,這需要進一步研究。

圖二 三大類 NAD+ 消耗酶。
A.一個 Sirtuins 使用煙酰胺腺嘌呤二核苷酸 (NAD+) 作為共底物從靶蛋白上的賴氨酸殘基中去除酰基。 NAD+ 被切割,產生煙酰胺 (NAM) 和 ADP-核糖,其中 ADP-核糖作為酰基受體,產生乙酰基-ADP-核糖 (乙酰基-ADPR)。
B.聚(ADP-核糖)聚合酶(PARP1-PARP3)使用 NAD+ 作為單(ADP-核糖)酯或聚(ADP-核糖)酯靶蛋白的共底物,產生 NAM 作為副產物。
C. NAD+ 糖水解酶和環狀 ADP-核糖 (cADPR) 合酶(CD38、CD157 和 SARM1)的反應。這組蛋白質的主要催化活性是 NAD+ 水解為 NAM 和 ADP-核糖。在較小程度上,CD38、CD157 和 SARM1 具有 ADP-核糖基環化酶活性,從 NAD+ 產生 NAM 和 cADPR。在酸性條件下,CD38 還可以進行鹼基交換反應,將 NAD(P)+ 的 NAM 交換為菸酸 (NA),生成菸酸腺嘌呤二核苷酸 (磷酸鹽) (NAAD(P))。據報導,CD38 和 CD157 能夠在其催化反應中使用替代底物。 CD38 可以將 NMN 降解為 NAM 和核糖一磷酸 (RMP),而 CD157 可以降解 NR,生成 NAM 和核糖 (R)。 NR,煙酰胺核苷。
A.一個 Sirtuins 使用煙酰胺腺嘌呤二核苷酸 (NAD+) 作為共底物從靶蛋白上的賴氨酸殘基中去除酰基。 NAD+ 被切割,產生煙酰胺 (NAM) 和 ADP-核糖,其中 ADP-核糖作為酰基受體,產生乙酰基-ADP-核糖 (乙酰基-ADPR)。
B.聚(ADP-核糖)聚合酶(PARP1-PARP3)使用 NAD+ 作為單(ADP-核糖)酯或聚(ADP-核糖)酯靶蛋白的共底物,產生 NAM 作為副產物。
C. NAD+ 糖水解酶和環狀 ADP-核糖 (cADPR) 合酶(CD38、CD157 和 SARM1)的反應。這組蛋白質的主要催化活性是 NAD+ 水解為 NAM 和 ADP-核糖。在較小程度上,CD38、CD157 和 SARM1 具有 ADP-核糖基環化酶活性,從 NAD+ 產生 NAM 和 cADPR。在酸性條件下,CD38 還可以進行鹼基交換反應,將 NAD(P)+ 的 NAM 交換為菸酸 (NA),生成菸酸腺嘌呤二核苷酸 (磷酸鹽) (NAAD(P))。據報導,CD38 和 CD157 能夠在其催化反應中使用替代底物。 CD38 可以將 NMN 降解為 NAM 和核糖一磷酸 (RMP),而 CD157 可以降解 NR,生成 NAM 和核糖 (R)。 NR,煙酰胺核苷。
PARPs 的消費
人類 PARP 蛋白家族由 17 種蛋白質組成,其特徵在於聚(ADP-核糖基)聚合酶或單(ADP-核糖基)聚合酶活性24。簡而言之,PARP 介導的 NAD+ 裂解會產生 NAM 和 ADP-核糖作為副產物,其中 ADP-核糖作為單一或共價連接的聚合物添加到 PARP 本身和其他受體蛋白中,這一過程稱為“聚 (ADP-核糖基化'(PARylation) (圖2b)。在所有 PARP 中,只有 PARP1、PARP2 和 PARP3 定位於細胞核(圖 1b)以響應早期 DNA 損傷,並在 DNA 損傷修復中起關鍵作用(補充框 2)。
PARP1 是該組中最典型的成員,它單獨負責大約 90% 的總 PARP 活性,至少響應 DNA 損傷。激活後,PARP1 與組蛋白和其他蛋白質一起 PARylate,這些蛋白質作為支架募集和激活其他 DNA 修復酶和蛋白質到病變部位以啟動 DNA 修復。由於高 PARP1 活性,DNA 損傷與大量 NAD+ 消耗有關。 PARP1 作為一種 NAD+ 響應信號分子,與衰老過程廣泛相關。然而,PARP1 是主要的 NAD+ 消費者之一,不僅在具有急性 DNA 損傷的細胞中,而且在正常和其他病理生理條件下也是如此,這支持了 PARP1 在調節 NAD+ 穩態中的關鍵作用。例如,在餵食高脂肪飲食的小鼠中,那些接受 PARP 抑製劑治療或缺乏 PARP1 和 PARP2 的小鼠的 NAD+ 水平增加,SIRT1 活性增加,線粒體功能改善,並且受到保護免受胰島素抵抗。在 A 組色素性乾皮病以及早衰症、共濟失調毛細血管擴張症和 Cockayne 綜合徵患者中觀察到 PARP1 激活、NAD+ 水平下降和 SIRT1 活性抑制之間的強相關性。值得注意的是,用 PARP1 抑製劑或 NAD+ 補充劑治療 Cockayne 綜合徵小鼠可延長壽命並改善由 PARP1 過度激活引起的嚴重表型,這提供了強有力的證據表明 PARP1 激活下游的負面後果是由響應廣泛 DNA 的 NAD+ 穩態失調介導的損傷和遺傳毒性應激。值得注意的是,PARP1 拮抗 SIRT1 活性的能力很可能是因為這些酶定位於同一個細胞區室——在這種情況下,是細胞核——並競爭同一個 NAD+ 池。然而,由於 PARP1 在 NAD+ 方面的 Km 和 Vmax 比 SIRT1 低,因此 PARP1 可能在 NAD+ 方面勝過 SIRT1,因為它具有更大的結合親和力和更快的動力學。
PARP2 在結構上與 PARP1 相關。它們共享幾個細胞過程所需的相似催化結構域,包括 DNA 修復和轉錄調節,並佔 PARP 活性的約 10%。 PARP2 活性也可能影響 NAD+ 生物利用度。與 Parp1 基因敲除小鼠一樣,Parp2 基因敲除小鼠表現出增強的 SIRT1 活性和改善的代謝功能,並且可以防止高脂飲食引起的肥胖。 PARP3 在 DNA 修復中也很重要,這表明 PARP1、PARP2 和 PARP3 中存在大量重疊和潛在冗餘。其他 PARPs (PARP4-PARP17) 在 NAD+ 穩態和細胞或器官中的全局代謝中的功能尚未完全確定,但它們被認為在調節細胞內 NAD+ 水平方面不太重要。總體而言,靶向 PARPs,特別是 PARP1,是衰老領域的一種有前途的治療策略。然而,需要更多的研究來充分了解 PARPs 對與年齡相關的 NAD+ 水平下降的貢獻。
CD38 和 CD157 消耗。
CD38 和 CD157 是具有糖水解酶和 ADP-核糖基環化酶活性的多功能胞外酶。 NAD+ 的糖水解是切割 NAD+ 內的糖苷鍵以產生 NAM 和 ADP-核糖的主要催化反應,而 ADP-核糖基環化酶活性產生環狀 ADP-核糖(圖 2c)。 CD38 還通過在酸性條件下將 NAD(P)+ 的 NAM 交換為 NA 並產生菸酸腺嘌呤二核苷酸(磷酸鹽)(NAAD(P))(圖 2c)來進行鹼基交換反應。值得注意的是,環狀 ADP-核糖、NAAD(P) 和 ADP-核糖都是調動 Ca2+ 的關鍵第二信使,說明 CD38 在激活 Ca2+ 信號傳導和調節基本細胞過程(例如免疫細胞活化、存活和代謝)中的關鍵作用。重要的是,除了 NAD+ 和 NADP+,NMN 正在成為 CD38 的替代底物 (REFS41,42),而 CD157 消耗 NR 作為替代底物 (圖 2c)。因此,用小分子抑製劑靶向 CD38 和 CD157 可以使這些常用的 NAD+ 前體代謝物更有效地恢復老年人的 NAD+ 水平。關於 CD157 酶促功能在細胞生物學或衰老中的作用知之甚少。然而,最近的證據表明,與 CD38 一樣,CD157 在衰老組織中上調並且可能在與衰老相關的疾病中發揮作用,例如類風濕性關節炎和癌症 46。
雖然 CD38 和 CD157 在基因上是同源的並且是同一酶家族的成員,但它們的結構、定位和在疾病中的作用不同。 CD38 是一種跨膜蛋白,具有 ii 型和/或 III 型取向,在 1970 年代後期被確定為 T 細胞活化標誌物,但現在已知其普遍表達,特別是在炎症期間。 CD157 是一種糖磷脂酰肌醇錨定蛋白,首先在造血系統的骨髓隔室中發現。然而,CD157 也由其他細胞表達,包括 B 細胞祖細胞、潘氏細胞和腸道、胰腺和腎臟中的內皮細胞。
除了酶促功能外,CD38 和 CD157 還充當細胞受體。例如,CD38 是一種粘附受體,與 CD31 相互作用以介導免疫細胞運輸和通過內皮細胞外滲。 CD38-CD31 軸似乎促進慢性淋巴細胞白血病淋巴細胞的增殖反應,表明 CD38 在血癌中的有害作用(見補充框 3)。然而,關於 CD38-CD31 相互作用的功能後果知之甚少。此外,CD38 被認為通過抗菌功能介導免疫,因為 Cd38 敲除小鼠的主要表型是它們無法對細菌產生免疫反應。 CD38 酶促功能是否對其抗菌功能是必需的以及這些功能在多大程度上依賴於 NAD+ 仍是未知數。然而,CD38 在抗菌素耐藥性中的作用似乎與從需要 NAD+ 才能存活和生長的細菌中隔離 NAD+ 或 NAD+ 相關代謝物有關。
CD157 作為受體的作用仍未完全探索。幾條證據表明,特定單克隆抗體對 CD157 的激活促進了中性粒細胞和單核細胞的運輸。此外,CD157 與整合素相互作用形成一種被癢病反應基因 1 蛋白 (SCRG1) 識別的受體,促進人間充質乾細胞的自我更新、遷移和成骨分化。然而,這些受體功能是否與 NAD+ 代謝相關仍不清楚。
SARM1 消耗
SARM1 僅在最近才與 CD38 和 CD157 一起被分配到 NAD+ 糖水解酶和環化酶家族。 SARM1 的酶活性(圖 2c)依賴於 Toll/白細胞介素受體(TIR)結構域,該結構域以前不知道具有催化活性並且通常參與蛋白質-蛋白質相互作用。 SARM1是否在生理條件下調節NAD+以及調節到何種程度仍然未知。然而,SARM1 介導的 NAD+ 降解在軸突損傷後的軸突變性中起關鍵作用(參見神經變性部分)。 SARM1 主要在神經元中表達並促進神經元形態發生和炎症;然而,SARM1 也由巨噬細胞和 T 淋巴細胞等免疫細胞表達,並調節它們的功能。 SARM1 最初是通過與 Toll 樣受體信號通路下游的 TRIF(含 TIR 結構域的銜接蛋白誘導干擾素-β)直接相互作用而被發現作為先天免疫反應的負調節因子。然而,SARM1 的免疫調節作用在很大程度上仍未得到表徵。儘管最初報導 SARM1 是巨噬細胞中趨化因子 CCL5 產生所必需的,但最近的證據表明 SARM1 在巨噬細胞中不表達,並且觀察到的趨化因子表型是由於 Sarm1 敲除小鼠品系的背景效應所致。儘管 SARM1 在免疫細胞中的作用存在爭議,但 SARM1 在軸突變性中發揮著無可爭議的關鍵作用,並且正在成為預防或改善神經退行性疾病和創傷性腦損傷的治療靶點(如神經退行性疾病部分所述)。
3. NAD+ 的細胞作用
超越 NAD+- 的主要群體
除了迄今為止討論的主要消耗 NAD+ 的酶組外,NAD+ 被廣泛用作生化反應的輔助因子或底物,有 300 多種酶依賴於 NAD+ 的活性。 因此,NAD+ 是關鍵細胞功能和適應代謝需求的介質。 其中一些關鍵的細胞過程包括代謝途徑、氧化還原穩態、維持和修復 DNA 以保障基因組穩定性、表觀遺傳調控和染色質重塑以及自噬。 總的來說,這些功能對於維持全身健康和體內平衡很重要。 然而,在衰老過程中,NAD+ 水平下降會影響這些過程並加劇與衰老相關的疾病(圖 3;補充框 2)
人類 PARP 蛋白家族由 17 種蛋白質組成,其特徵在於聚(ADP-核糖基)聚合酶或單(ADP-核糖基)聚合酶活性24。簡而言之,PARP 介導的 NAD+ 裂解會產生 NAM 和 ADP-核糖作為副產物,其中 ADP-核糖作為單一或共價連接的聚合物添加到 PARP 本身和其他受體蛋白中,這一過程稱為“聚 (ADP-核糖基化'(PARylation) (圖2b)。在所有 PARP 中,只有 PARP1、PARP2 和 PARP3 定位於細胞核(圖 1b)以響應早期 DNA 損傷,並在 DNA 損傷修復中起關鍵作用(補充框 2)。
PARP1 是該組中最典型的成員,它單獨負責大約 90% 的總 PARP 活性,至少響應 DNA 損傷。激活後,PARP1 與組蛋白和其他蛋白質一起 PARylate,這些蛋白質作為支架募集和激活其他 DNA 修復酶和蛋白質到病變部位以啟動 DNA 修復。由於高 PARP1 活性,DNA 損傷與大量 NAD+ 消耗有關。 PARP1 作為一種 NAD+ 響應信號分子,與衰老過程廣泛相關。然而,PARP1 是主要的 NAD+ 消費者之一,不僅在具有急性 DNA 損傷的細胞中,而且在正常和其他病理生理條件下也是如此,這支持了 PARP1 在調節 NAD+ 穩態中的關鍵作用。例如,在餵食高脂肪飲食的小鼠中,那些接受 PARP 抑製劑治療或缺乏 PARP1 和 PARP2 的小鼠的 NAD+ 水平增加,SIRT1 活性增加,線粒體功能改善,並且受到保護免受胰島素抵抗。在 A 組色素性乾皮病以及早衰症、共濟失調毛細血管擴張症和 Cockayne 綜合徵患者中觀察到 PARP1 激活、NAD+ 水平下降和 SIRT1 活性抑制之間的強相關性。值得注意的是,用 PARP1 抑製劑或 NAD+ 補充劑治療 Cockayne 綜合徵小鼠可延長壽命並改善由 PARP1 過度激活引起的嚴重表型,這提供了強有力的證據表明 PARP1 激活下游的負面後果是由響應廣泛 DNA 的 NAD+ 穩態失調介導的損傷和遺傳毒性應激。值得注意的是,PARP1 拮抗 SIRT1 活性的能力很可能是因為這些酶定位於同一個細胞區室——在這種情況下,是細胞核——並競爭同一個 NAD+ 池。然而,由於 PARP1 在 NAD+ 方面的 Km 和 Vmax 比 SIRT1 低,因此 PARP1 可能在 NAD+ 方面勝過 SIRT1,因為它具有更大的結合親和力和更快的動力學。
PARP2 在結構上與 PARP1 相關。它們共享幾個細胞過程所需的相似催化結構域,包括 DNA 修復和轉錄調節,並佔 PARP 活性的約 10%。 PARP2 活性也可能影響 NAD+ 生物利用度。與 Parp1 基因敲除小鼠一樣,Parp2 基因敲除小鼠表現出增強的 SIRT1 活性和改善的代謝功能,並且可以防止高脂飲食引起的肥胖。 PARP3 在 DNA 修復中也很重要,這表明 PARP1、PARP2 和 PARP3 中存在大量重疊和潛在冗餘。其他 PARPs (PARP4-PARP17) 在 NAD+ 穩態和細胞或器官中的全局代謝中的功能尚未完全確定,但它們被認為在調節細胞內 NAD+ 水平方面不太重要。總體而言,靶向 PARPs,特別是 PARP1,是衰老領域的一種有前途的治療策略。然而,需要更多的研究來充分了解 PARPs 對與年齡相關的 NAD+ 水平下降的貢獻。
CD38 和 CD157 消耗。
CD38 和 CD157 是具有糖水解酶和 ADP-核糖基環化酶活性的多功能胞外酶。 NAD+ 的糖水解是切割 NAD+ 內的糖苷鍵以產生 NAM 和 ADP-核糖的主要催化反應,而 ADP-核糖基環化酶活性產生環狀 ADP-核糖(圖 2c)。 CD38 還通過在酸性條件下將 NAD(P)+ 的 NAM 交換為 NA 並產生菸酸腺嘌呤二核苷酸(磷酸鹽)(NAAD(P))(圖 2c)來進行鹼基交換反應。值得注意的是,環狀 ADP-核糖、NAAD(P) 和 ADP-核糖都是調動 Ca2+ 的關鍵第二信使,說明 CD38 在激活 Ca2+ 信號傳導和調節基本細胞過程(例如免疫細胞活化、存活和代謝)中的關鍵作用。重要的是,除了 NAD+ 和 NADP+,NMN 正在成為 CD38 的替代底物 (REFS41,42),而 CD157 消耗 NR 作為替代底物 (圖 2c)。因此,用小分子抑製劑靶向 CD38 和 CD157 可以使這些常用的 NAD+ 前體代謝物更有效地恢復老年人的 NAD+ 水平。關於 CD157 酶促功能在細胞生物學或衰老中的作用知之甚少。然而,最近的證據表明,與 CD38 一樣,CD157 在衰老組織中上調並且可能在與衰老相關的疾病中發揮作用,例如類風濕性關節炎和癌症 46。
雖然 CD38 和 CD157 在基因上是同源的並且是同一酶家族的成員,但它們的結構、定位和在疾病中的作用不同。 CD38 是一種跨膜蛋白,具有 ii 型和/或 III 型取向,在 1970 年代後期被確定為 T 細胞活化標誌物,但現在已知其普遍表達,特別是在炎症期間。 CD157 是一種糖磷脂酰肌醇錨定蛋白,首先在造血系統的骨髓隔室中發現。然而,CD157 也由其他細胞表達,包括 B 細胞祖細胞、潘氏細胞和腸道、胰腺和腎臟中的內皮細胞。
除了酶促功能外,CD38 和 CD157 還充當細胞受體。例如,CD38 是一種粘附受體,與 CD31 相互作用以介導免疫細胞運輸和通過內皮細胞外滲。 CD38-CD31 軸似乎促進慢性淋巴細胞白血病淋巴細胞的增殖反應,表明 CD38 在血癌中的有害作用(見補充框 3)。然而,關於 CD38-CD31 相互作用的功能後果知之甚少。此外,CD38 被認為通過抗菌功能介導免疫,因為 Cd38 敲除小鼠的主要表型是它們無法對細菌產生免疫反應。 CD38 酶促功能是否對其抗菌功能是必需的以及這些功能在多大程度上依賴於 NAD+ 仍是未知數。然而,CD38 在抗菌素耐藥性中的作用似乎與從需要 NAD+ 才能存活和生長的細菌中隔離 NAD+ 或 NAD+ 相關代謝物有關。
CD157 作為受體的作用仍未完全探索。幾條證據表明,特定單克隆抗體對 CD157 的激活促進了中性粒細胞和單核細胞的運輸。此外,CD157 與整合素相互作用形成一種被癢病反應基因 1 蛋白 (SCRG1) 識別的受體,促進人間充質乾細胞的自我更新、遷移和成骨分化。然而,這些受體功能是否與 NAD+ 代謝相關仍不清楚。
SARM1 消耗
SARM1 僅在最近才與 CD38 和 CD157 一起被分配到 NAD+ 糖水解酶和環化酶家族。 SARM1 的酶活性(圖 2c)依賴於 Toll/白細胞介素受體(TIR)結構域,該結構域以前不知道具有催化活性並且通常參與蛋白質-蛋白質相互作用。 SARM1是否在生理條件下調節NAD+以及調節到何種程度仍然未知。然而,SARM1 介導的 NAD+ 降解在軸突損傷後的軸突變性中起關鍵作用(參見神經變性部分)。 SARM1 主要在神經元中表達並促進神經元形態發生和炎症;然而,SARM1 也由巨噬細胞和 T 淋巴細胞等免疫細胞表達,並調節它們的功能。 SARM1 最初是通過與 Toll 樣受體信號通路下游的 TRIF(含 TIR 結構域的銜接蛋白誘導干擾素-β)直接相互作用而被發現作為先天免疫反應的負調節因子。然而,SARM1 的免疫調節作用在很大程度上仍未得到表徵。儘管最初報導 SARM1 是巨噬細胞中趨化因子 CCL5 產生所必需的,但最近的證據表明 SARM1 在巨噬細胞中不表達,並且觀察到的趨化因子表型是由於 Sarm1 敲除小鼠品系的背景效應所致。儘管 SARM1 在免疫細胞中的作用存在爭議,但 SARM1 在軸突變性中發揮著無可爭議的關鍵作用,並且正在成為預防或改善神經退行性疾病和創傷性腦損傷的治療靶點(如神經退行性疾病部分所述)。
3. NAD+ 的細胞作用
超越 NAD+- 的主要群體
除了迄今為止討論的主要消耗 NAD+ 的酶組外,NAD+ 被廣泛用作生化反應的輔助因子或底物,有 300 多種酶依賴於 NAD+ 的活性。 因此,NAD+ 是關鍵細胞功能和適應代謝需求的介質。 其中一些關鍵的細胞過程包括代謝途徑、氧化還原穩態、維持和修復 DNA 以保障基因組穩定性、表觀遺傳調控和染色質重塑以及自噬。 總的來說,這些功能對於維持全身健康和體內平衡很重要。 然而,在衰老過程中,NAD+ 水平下降會影響這些過程並加劇與衰老相關的疾病(圖 3;補充框 2)

圖三 衰老過程中的 NAD+ 代謝。
A.一個降低的煙酰胺腺嘌呤二核苷酸 (NAD+) 水平與衰老相關的各種過程有關(另見補充框 2)。
A1衰老與異常的促炎免疫細胞激活或“炎症”有關,導致持續的低度炎症。這部分是由衰老細胞的積累引起的,衰老細胞通過衰老相關分泌表型 (SASP) 促進巨噬細胞表型極化向促炎 M1 狀態,從而驅動炎症。有證據表明,響應這些巨噬細胞中的 SASP,消耗 NAD+ 的酶 CD38 和聚(ADP-核糖)聚合酶 (PARPs) 的表達增加,導致 NAD+ 水平下降,並且這些機制對 NAD+ 的降低有重要作用老化水平。此外,已經表明,在衰老的 T 細胞中,線粒體功能下降,這導致促炎細胞因子的分泌增加,這些細胞因子促進炎症狀態並誘導衰老。
A2.抗體軸突變性是許多與年齡相關的神經元疾病的前兆,其特徵是 NAD+ 快速消耗。在正常生理條件下,NAD+ 生物合成酶、煙酰胺單核苷酸腺苷酸轉移酶 (NMNAT) 對軸突變性具有保護作用,它們的表達支持軸突的維持並防止神經變性。特別是,NMNAT2 是軸突中一個重要的生存因子,它需要不斷地從體細胞(它被合成的地方)輸送到它,以解釋它的快速周轉,並且這些運輸過程在軸突退化期間受到干擾。此外,消耗 NAD+ 的酶 SARM1 被軸突損傷激活,並通過促進 NAD+ 降解介導軸突變性。
A3.交流自噬是一個關鍵的細胞分解代謝過程,它使細胞能夠適應可變的營養供應並用於細胞質量控制,從而去除有缺陷的細胞器和蛋白質。自噬通過 sirtuins(主要是 SIRT1)在 NAD+ 水平的下游進行調節。 NAD+ 水平的下降會降低整體自噬通量以及通過線粒體自噬選擇性去除線粒體,這表明自噬缺陷可能是衰老過程中 NAD+ 耗盡的結果,從而導致細胞功能障礙。
B.因為 NAD+ 是各種酶的輔助因子,NAD+ 的丟失會影響過多的細胞過程。例如,NAD+ 是 SIRT1 等表觀遺傳調節因子的活性所必需的,其水平下降會導致組蛋白修飾發生變化,從而影響染色質組織和基因表達中的功能。有證據表明,與衰老相關的 NAD+ 缺失與 PARPs 表達增加有關,這可能是由於 DNA 損傷水平增加和衰老過程中需要 DNA 修復引起的(圖 Ba)。 NAD+ 還影響核心時鐘成分 CLOCK 和 BMAL 的轉錄活性,從而調節重要代謝基因以及煙酰胺磷酸核糖基轉移酶 (NAMPT) 的晝夜節律表達,而 NAMPT 又是 NAD+ 水平晝夜節律振盪所必需的(圖 Bb)。 NAD+ 水平降低還會干擾 PARP 和 sirtuin 在 DNA 修復中的活性,導致基因組不穩定:衰老和癌症的標誌(Bc 組)。 ADPR,ADP-核糖; ATG,自噬相關蛋白; FOXO,叉頭盒蛋白 O; ROS,活性氧。
A.一個降低的煙酰胺腺嘌呤二核苷酸 (NAD+) 水平與衰老相關的各種過程有關(另見補充框 2)。
A1衰老與異常的促炎免疫細胞激活或“炎症”有關,導致持續的低度炎症。這部分是由衰老細胞的積累引起的,衰老細胞通過衰老相關分泌表型 (SASP) 促進巨噬細胞表型極化向促炎 M1 狀態,從而驅動炎症。有證據表明,響應這些巨噬細胞中的 SASP,消耗 NAD+ 的酶 CD38 和聚(ADP-核糖)聚合酶 (PARPs) 的表達增加,導致 NAD+ 水平下降,並且這些機制對 NAD+ 的降低有重要作用老化水平。此外,已經表明,在衰老的 T 細胞中,線粒體功能下降,這導致促炎細胞因子的分泌增加,這些細胞因子促進炎症狀態並誘導衰老。
A2.抗體軸突變性是許多與年齡相關的神經元疾病的前兆,其特徵是 NAD+ 快速消耗。在正常生理條件下,NAD+ 生物合成酶、煙酰胺單核苷酸腺苷酸轉移酶 (NMNAT) 對軸突變性具有保護作用,它們的表達支持軸突的維持並防止神經變性。特別是,NMNAT2 是軸突中一個重要的生存因子,它需要不斷地從體細胞(它被合成的地方)輸送到它,以解釋它的快速周轉,並且這些運輸過程在軸突退化期間受到干擾。此外,消耗 NAD+ 的酶 SARM1 被軸突損傷激活,並通過促進 NAD+ 降解介導軸突變性。
A3.交流自噬是一個關鍵的細胞分解代謝過程,它使細胞能夠適應可變的營養供應並用於細胞質量控制,從而去除有缺陷的細胞器和蛋白質。自噬通過 sirtuins(主要是 SIRT1)在 NAD+ 水平的下游進行調節。 NAD+ 水平的下降會降低整體自噬通量以及通過線粒體自噬選擇性去除線粒體,這表明自噬缺陷可能是衰老過程中 NAD+ 耗盡的結果,從而導致細胞功能障礙。
B.因為 NAD+ 是各種酶的輔助因子,NAD+ 的丟失會影響過多的細胞過程。例如,NAD+ 是 SIRT1 等表觀遺傳調節因子的活性所必需的,其水平下降會導致組蛋白修飾發生變化,從而影響染色質組織和基因表達中的功能。有證據表明,與衰老相關的 NAD+ 缺失與 PARPs 表達增加有關,這可能是由於 DNA 損傷水平增加和衰老過程中需要 DNA 修復引起的(圖 Ba)。 NAD+ 還影響核心時鐘成分 CLOCK 和 BMAL 的轉錄活性,從而調節重要代謝基因以及煙酰胺磷酸核糖基轉移酶 (NAMPT) 的晝夜節律表達,而 NAMPT 又是 NAD+ 水平晝夜節律振盪所必需的(圖 Bb)。 NAD+ 水平降低還會干擾 PARP 和 sirtuin 在 DNA 修復中的活性,導致基因組不穩定:衰老和癌症的標誌(Bc 組)。 ADPR,ADP-核糖; ATG,自噬相關蛋白; FOXO,叉頭盒蛋白 O; ROS,活性氧。
4. NAD+ 依賴性機制
在衰老過程中,NAD+ 水平下降,許多與 NAD+ 降解和生物合成相關的酶發生改變。 NAD+ 與衰老的 10 個標誌之間的關係得到了廣泛的審查 (另見補充表 1)。此外,衰老過程中 NAD+ 水平的下降與衰老相關疾病的發展和進展有關,包括動脈粥樣硬化、關節炎、高血壓、認知能力下降、糖尿病和癌症。在本節中,我們將重點關注影響或受衰老影響的主要細胞過程,例如代謝功能障礙、DNA 修復失敗和基因組不穩定性、炎症、細胞衰老和神經退行性變,並討論它們受 NAD+ 水平的調節。所有這些過程以及與之相關的與年齡相關的疾病都可能受益於 NAD+ 的補充。使用膳食前體恢復 NAD+ 水平並使用小分子抑製劑靶向 NAD+ 降解酶已成為恢復 NAD+ 水平的潛在治療策略,從而為緩解與年齡相關的衰退和疾病提供機會(圖 4)(另見下一節)。
在衰老過程中,NAD+ 水平下降,許多與 NAD+ 降解和生物合成相關的酶發生改變。 NAD+ 與衰老的 10 個標誌之間的關係得到了廣泛的審查 (另見補充表 1)。此外,衰老過程中 NAD+ 水平的下降與衰老相關疾病的發展和進展有關,包括動脈粥樣硬化、關節炎、高血壓、認知能力下降、糖尿病和癌症。在本節中,我們將重點關注影響或受衰老影響的主要細胞過程,例如代謝功能障礙、DNA 修復失敗和基因組不穩定性、炎症、細胞衰老和神經退行性變,並討論它們受 NAD+ 水平的調節。所有這些過程以及與之相關的與年齡相關的疾病都可能受益於 NAD+ 的補充。使用膳食前體恢復 NAD+ 水平並使用小分子抑製劑靶向 NAD+ 降解酶已成為恢復 NAD+ 水平的潛在治療策略,從而為緩解與年齡相關的衰退和疾病提供機會(圖 4)(另見下一節)。
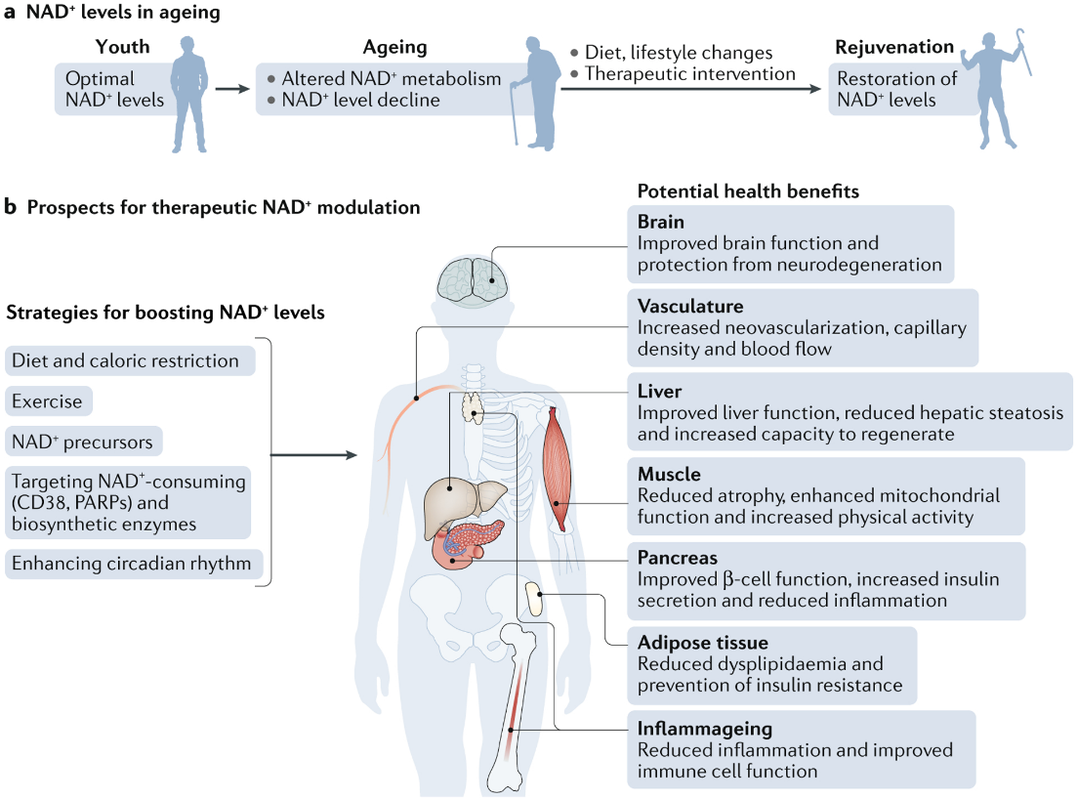
圖四 恢復 NAD+ 水平的治療方法及其對健康的影響。
衰老與促進或加劇衰老相關疾病的煙酰胺腺嘌呤二核苷酸 (NAD+) 水平降低有關。因此,恢復 NAD+ 水平已成為預防和治療與衰老相關的疾病以及在衰老過程中恢復健康和活力的一種治療方法(a 部分)。提高 NAD+ 水平的一些潛在策略包括改變生活方式,例如增加鍛煉、減少熱量攝入、健康飲食以及通過遵循健康的睡眠習慣和進餐時間來遵循一致的每日晝夜節律模式。另一種方法是使用小分子抑製劑或激活劑來促進 NAD+ 生物合成和使用膳食補充劑,包括 NAD+ 前體,例如煙酰胺單核苷酸和煙酰胺核苷。所有這些方法都可以促進組織 NAD+ 水平,對健康有益。這些包括改善組織和器官功能、防止認知衰退、改善代謝健康、減少炎症和增加生理益處,例如增加身體活動,這可能共同延長患者的健康壽命和潛在的壽命(b 部分)。
衰老與促進或加劇衰老相關疾病的煙酰胺腺嘌呤二核苷酸 (NAD+) 水平降低有關。因此,恢復 NAD+ 水平已成為預防和治療與衰老相關的疾病以及在衰老過程中恢復健康和活力的一種治療方法(a 部分)。提高 NAD+ 水平的一些潛在策略包括改變生活方式,例如增加鍛煉、減少熱量攝入、健康飲食以及通過遵循健康的睡眠習慣和進餐時間來遵循一致的每日晝夜節律模式。另一種方法是使用小分子抑製劑或激活劑來促進 NAD+ 生物合成和使用膳食補充劑,包括 NAD+ 前體,例如煙酰胺單核苷酸和煙酰胺核苷。所有這些方法都可以促進組織 NAD+ 水平,對健康有益。這些包括改善組織和器官功能、防止認知衰退、改善代謝健康、減少炎症和增加生理益處,例如增加身體活動,這可能共同延長患者的健康壽命和潛在的壽命(b 部分)。
4.1. 代謝功能障礙
肥胖是一種日益嚴重的全球流行病。肥胖者更容易患上以肥胖增加、胰島素抵抗、高血糖、高血壓和血脂異常為特徵的代謝疾病。因此,這些人患 2 型糖尿病、心血管疾病、非酒精性脂肪肝、動脈粥樣硬化、中風和癌症的風險更高。因此,肥胖會加速和加劇衰老,並與壽命縮短有關。在過去的二十年中,越來越多的證據表明,靶向 NAD+ 代謝可能會提供治療益處並有助於治療代謝疾病和衰老。
NAD+ 是通過調節酵母提取物的代謝率首次發現的,因此 NAD+ 與代謝之間的聯繫已為人所知近一個世紀。我們現在知道 NAD+ 位於新陳代謝的核心並調節多種代謝途徑的代謝通量(詳情參見框 1 和補充框 2)。因此,NAD+ 穩態是各種代謝組織正常功能所必需的,包括脂肪、肌肉、腸、腎和肝。然而,最近在釀酒酵母中發現了另一個關鍵發現,表明長壽蛋白 Sir2(酵母 sirtuin)以 NAD+ 依賴性方式表現出其延長壽命的作用。這一突破性發現表明 Sir2 的活性可能與代謝狀態有關。為了支持這一假設,現在已經在哺乳動物系統中證明了延長壽命的代謝操作,例如運動、熱量限制限時餵養和生酮飲食,以及健康的晝夜節律(包括定期睡眠模式 80),部分通過增加 NAD+ 水平起作用,這會導致去乙酰化酶的激活。
改變或破壞代謝狀態,例如高脂飲食、產後體重減輕和晝夜節律紊亂的結果,可導致 NAD+ 水平降低,從而降低去乙酰化酶和其他 NAD+ 的活性依賴的細胞過程(詳見補充框 2)。相反,最近顯示增加細胞 NAD+ 水平可以減少還原性壓力並推動代謝反應的方向性。此外,較高的 NAD+ 水平可以促進核 SIRT1 和線粒體 SIRT3 的去乙酰化酶和去乙酰化酶活性,從而調節線粒體功能並防止高脂飲食引起的代謝疾病。
總之,這些關鍵研究提供了強有力的證據和基本原理,即靶向 NAD+ 降解途徑或提高 NAD+ 水平可以影響代謝過程並可以預防代謝疾病。為了進一步支持該模型,許多研究現在表明,包括 Parp1 敲除和 Cd38 敲除小鼠在內的小鼠模型,或用 PARP 或 CD38 抑製劑治療的小鼠,具有超生理水平的 NAD+。它們還可以防止肥胖,提高代謝率,並且在高脂肪飲食條件和衰老過程中(相對)正常的葡萄糖代謝。此外,接受高脂肪飲食的小鼠炎症增加,導致 NAMPT 表達降低,NAD+ 補救途徑的活性降低 82 (BOX 1),為肥胖期間 NAD+ 水平下降的原因提供了潛在的機制解釋。根據這一機制,脂肪細胞特異性缺失 NAMPT 的小鼠其脂肪組織中的 NAD+ 水平較低,胰島素抵抗增加和代謝功能障礙增加,這可以通過補充 NMN 來挽救。此外,最近的多項研究表明,可以恢復與衰老相關的低 NAD+ 水平的 NAD+ 補充劑(NR 和 NMN)在囓齒動物模型中也可以預防肥胖,這表明這些補充劑可以作為治療方法來恢復人類肥胖患者的代謝健康。最近的幾項臨床試驗已經開始研究 NAD+ 前體在人類肥胖患者中改善代謝健康和葡萄糖代謝的功效。到目前為止,大多數這些研究都是在健康個體中完成的(見表 1),以測試可以安全提高 NAD+ 水平的 NR 和 NMN 的有效劑量。然而,最近的兩項隨機雙盲研究使用 NR 治療超重和肥胖患者 6 周和 12 週 。不幸的是,雖然 NR 可以有效增加這些人的 NAD+ 水平,但兩項研究的參與者都沒有表現出任何體重減輕、胰島素敏感性增加或線粒體功能增強等代謝參數的跡象。因此,目前尚不清楚靶向 NAD+ 代謝是否能有效治療肥胖或老年人的代謝疾病。
4.2.免疫細胞功能失調
炎症現在被認為是衰老的標誌和疾病的關鍵驅動因素(圖 3Aa),包括與衰老相關的疾病,並被描述為發病和死亡的主要風險因素。慢性炎症通過免疫細胞和代謝細胞(例如肝細胞和脂肪細胞)之間的複雜串擾,影響葡萄糖和脂質攝取以及胰島素敏感性等代謝過程,對全身代謝產生深遠影響。儘管在過去十年中人們對免疫代謝的興趣增加了,但人們對 NAD+ 如何影響慢性炎症和免疫細胞功能知之甚少。接下來我們將討論正在出現的畫面。
先天免疫。
慢性低度炎症,特徵為先天免疫系統異常激活,促炎細胞因子(如腫瘤壞死因子 (TNF)、IL-6 和 IL-1β)表達增強,以及免疫複合物(如 NLRP3 炎症小體)激活,現在被認為是衰老相關疾病和代謝疾病的關鍵驅動因素。此外,現在發現改變的巨噬細胞活化和表型極化是這種炎症的關鍵來源。例如,在內臟脂肪等肥胖組織中,外周單核細胞被受壓的脂肪細胞募集,導致促炎性 M1 樣巨噬細胞的進行性浸潤和駐留的抗炎 M2 巨噬細胞的移位。內臟脂肪中巨噬細胞極化狀態的這種轉變伴隨著促炎細胞因子的表達增強、胰島素抵抗和脂肪分解率的降低。 100 多年前,巨噬細胞的發現者 Elie Metchnikoff 是第一個觀察到衰老組織中巨噬細胞豐度增加的人。儘管這一觀察最初被忽視,但現在越來越多的證據表明,衰老不僅會導致巨噬細胞豐度增加,而且還會伴隨巨噬細胞極化狀態和功能的改變,這正在成為炎症的關鍵驅動因素。
一些表明 NAD+ 影響巨噬細胞功能的最早研究表明,抑制 NAMPT 和隨後消耗巨噬細胞中的 NAD+ 池會降低促炎細胞因子(例如 TNF)的分泌,並導致形態學變化,例如減少這些細胞中的擴散。最近的研究表明,NAD+ 是巨噬細胞功能的關鍵調節因子,巨噬細胞活化與 NAD+ 生物合成或降解途徑的上調有關,這取決於獲得的命運。例如,我們的實驗室最近表明,促炎 (M1) 巨噬細胞極化與 CD38 表達增強有關,從而導致 NAD+ 消耗增加。相反,抗炎 (M2) 巨噬細胞極化與依賴於 NAMPT45 的 NAD+ 水平的增加有關。阻斷 M1 巨噬細胞和 M2 巨噬細胞中的 NAM 補救途徑 (BOX 1) 顯著降低了與 M1 和 M2 表型相關的選定基因的基因表達。這種效果可以通過補充 NAD+ 前體 NMN 和 NR 來挽救,它們繞過和挽救 NAMPT 抑制。與 M1 巨噬細胞相比,M2 巨噬細胞需要更多的 NR/NMN 來拯救巨噬細胞活化,這表明 NAD+ 是一般巨噬細胞活化的關鍵代謝物,其代謝受到差異調節以控制 M1 和 M2 巨噬細胞中不同的生物過程和功能。
與我們的結果一致,最近的一項研究發現,M1 巨噬細胞極化與 NAD+ 的降解增強有關,抑制 NAMPT 可阻斷 M1 巨噬細胞的糖酵解轉變,限制體外促炎反應並減少體內對膿毒症的全身炎症反應。這項研究和最近的另一份報告表明,這種 NAD+ 轉換,特別是在巨噬細胞 M1 極化的最初幾個小時內,取決於活性氧誘導的 DNA 損傷和 PARP1 的激活。然而,我們實驗室最近發表的結果沒有檢測到在 M1 巨噬細胞極化期間 DNA 損傷或 PARP1 激活的證據。相比之下,我們的結果表明 CD38 是 M1 巨噬細胞中主要的 NAD+ 消耗酶。這些結果與最近的工作一致,表明 M1 巨噬細胞免受活性氧誘導的 DNA 損傷,這是由於參與抗氧化防禦的基因轉錄增加,如 SOD2 (REFS102-104)。因此,這些相互矛盾的觀察強調需要更好地描述促炎和抗炎巨噬細胞極化過程中 NAD+ 的主要消費者,以確定觀察到的 NAD+ 水平下降是否取決於背景和時間,並確定 NAD+ 水平影響促炎的分子機制和抗炎基因表達和巨噬細胞功能。
在衰老過程中,NAD+ 水平下降與肝臟和脂肪中促炎性 M1 樣常駐巨噬細胞的積累增加有關,其特點是 CD38 表達增加和 NADase 活性升高。通過體外和體內方法,發現這些過表達 CD38 的 M1 樣巨噬細胞直接被衰老細胞分泌的炎性細胞因子激活(稍後將詳細討論)。此外,衰老巨噬細胞的特徵是 NAD+ 從頭合成受損,這本身可能會影響衰老過程中的巨噬細胞功能6。
關於炎症,上述研究表明促炎 M1 樣巨噬細胞(除了衰老細胞;見下文)可能是衰老組織中促炎細胞因子的主要來源。衰老與主要由髓系免疫細胞表達的 NLRP3 炎症小體的激活增加相關,以及隨後 IL-1β 的表達增加,IL-1β 是與促進衰老相關疾病有關的主要細胞因子。促炎細胞因子的表達增強可能會導致炎症的惡性循環,導致更大的炎症、增強的組織和 DNA 損傷、進一步激活 NAD+ 的主要消費者,如 CD38 和 PARPs,並加速與年齡相關的生理衰退。
因此,靶向巨噬細胞免疫代謝途徑,特別是那些調節 NAD+ 生物合成或降解途徑的途徑,可以作為激活或抑制巨噬細胞功能和調節巨噬細胞極化狀態的治療策略進行探索。這當然與調節炎症相關,但也可以用於緩解由慢性炎症驅動的疾病,例如神經退行性疾病和自身炎症性疾病,也可以作為癌症的治療策略,其中巨噬細胞極化可以是腫瘤促進 (M2) 或腫瘤抑制性(M1)
適應性免疫。
與先天免疫系統一樣,衰老的特徵是由於適應性免疫細胞的功能改變(稱為免疫衰老)而導致建立有效適應性免疫反應的能力降低。
衰老導致免疫細胞群失衡或傾斜,包括初始 T 細胞和 B 細胞水平降低、T 細胞抗原受體多樣性喪失和虛擬記憶 T 細胞水平增加。 NAD+ 和 NAD+ 消耗酶在 T 細胞生物學中的調節作用已得到證實;然而,它們對適應性免疫老化的貢獻在很大程度上是未知的。一方面,細胞外 NAD+ 被認為是一種危險信號,可導致特定 T 細胞亞群(例如調節性 T 細胞)中的細胞死亡。另一方面,NAD+ 似乎表現出免疫調節特性,例如影響 T 細胞極化 。然而,NAD+ 是否促進特定的 T 細胞表型以及用 NAD+ 前體操縱 NAD+ 代謝是否會導致類似的免疫調節特性仍然未知。
適應性免疫衰老的一個既定標誌是高細胞毒性 CD8+CD28-T 細胞的記憶群體的擴展,其特徵是效應分子(如粒酶 B115)的高分泌。該細胞群的特點是 SIRT1 和 FOXO1 水平降低,從而導致糖酵解能力和顆粒酶 B 產量增強。這些研究強調了通過操縱與年齡相關的適應性免疫功能障礙中的 NAD+ 相關途徑進行代謝重編程的潛力。通過 CD38 抑制上調 NAD+-SIRT1-FOXO1 軸可增加 T 輔助細胞 1 和 17 雜合細胞的效應功能,未來使用 CD38 抑製劑或 NAD+ 前體的研究可能顯示靶向 NAD+ 在衰老適應性免疫系統中的治療潛力。
適應性免疫衰老的另一個免疫學特徵是耗盡的 T 細胞數量增加,其特徵是抑制性受體分子(例如 PD1 和 TIM3)的表達、增殖能力降低和效應功能降低 118,119。 PD1 是免疫檢查點的一個組成部分,其阻斷通常用作抗癌策略,但也有人提出恢復老化 T 細胞的效應功能。關於 NAD+ 代謝,最近的一項研究表明,CD38 過表達與 PD1 阻斷抗性癌症中功能失調和耗竭的 CD8 T 群體相關 123,124,突出了將 CD38 抑制研究擴展到與年齡相關的耗竭 T 細胞的潛力。然而,儘管非常有趣,但這一假設仍有待深入探討,需要更多的研究來確定這種方法的臨床前療效。
因此,總體而言,需要做更多的工作來確定操縱 NAD+ 水平是否能有效逆轉適應性免疫系統中與衰老相關的免疫功能障礙,同樣重要的是,這種操縱是否安全。
4.3. 細胞衰老
在衰老過程中,暴露於代謝、基因毒性或致癌基因誘導的壓力的細胞經歷了本質上不可逆的細胞週期停滯,稱為細胞衰老。衰老細胞的一種主要表型以及它們如何促進疾病是炎症介質(主要是細胞因子和趨化因子)的表達增加,稱為衰老相關分泌表型 (SASP),它通過乾擾幹細胞導致組織穩態受損再生、組織和傷口修復和炎症(圖 3Aa)。隨著衰老細胞的數量隨著年齡的增長而逐漸增加,細胞衰老與幾種與年齡相關的疾病有關,用藥物抗衰老劑清除衰老細胞可能是治療幾種以前無法治癒的疾病的有效方法,包括阿爾茨海默病。此外,旨在提高衰老過程中細胞 NAD+ 水平的治療是延長健康壽命的有希望的目標,但 NAD+ 如何影響細胞衰老尚不清楚。最近表明,衰老細胞上調了 NAM 補救酶 NAMPT(BOX 1)的表達,並且衰老細胞的 SASP 取決於 NAD+ 水平。用 NMN 治療衰老細胞可以提高 SASP,導致慢性炎症增加,並可以促進炎症驅動癌症的發展。這些發現表明,服用 NAD+ 增強補充劑,如 NR 和 NMN,可能會以長期副作用為代價,如增強慢性炎症和癌症發展。因此,更好地了解提高 NAD+ 水平的益處和無人關注的副作用將是未來研究和正在進行的臨床試驗的一個重要重點領域。由於炎症是一個非常複雜和多用途的過程,需要更多的研究來更好地了解 NAD+ 水平如何影響不同的炎症狀態以及在什麼情況下,並確定 NAD+ 代謝如何從機制上影響炎症免疫和衰老細胞的生物學。
儘管有充分證據表明衰老組織中衰老細胞的積累以及這些組織中 NAD+ 水平的下降,但沒有研究將炎症性衰老細胞的積累與衰老過程中的 NAD+ 水平聯繫起來。最近,研究表明哺乳動物組織中的 CD38 水平隨著年齡的增長而增加,並且 CD38 被認為是導致衰老過程中 NAD+ 水平下降的主要 NAD+ 消耗酶。然而,驅動衰老組織中 CD38 表達增加的機制以及哪些細胞在這些組織中表達 CD38 尚不清楚。最近的觀察表明,先天免疫細胞,尤其是巨噬細胞,可能是響應 SASP 並伴有 NAD+ 降解的主要細胞群,從而導致整個生物體的 NAD+ 水平下降。我們發現,在來自衰老細胞的條件培養基中共培養或暴露於條件培養基中的巨噬細胞增強了消耗 NAD+ 的酶 CD38 的表達並增加了增殖。重要的是,另一組也獨立地表明,衰老細胞及其 SASP 激活 CD38 表達並促進巨噬細胞中 CD38 依賴性 NADase 活性。此外,我們在衰老小鼠模型和用誘導衰老的化學治療試劑阿黴素治療的小鼠中發現,衰老細胞在代謝組織(例如內臟脂肪和肝臟)中的積累可以直接激活組織駐留巨噬細胞中 CD38 的表達。為了進一步支持炎症、衰老細胞負荷和 NAD+ 之間的聯繫,最近在小鼠身上進行的一項研究還表明,線粒體功能失調的細胞會啟動促炎程序,並分泌促炎細胞因子。這與更大的衰老細胞負擔、增加的代謝和身體功能障礙以及過早衰老有關。在這種情況下,補充 NR 能夠部分挽救這種多病綜合徵,部分是通過減少炎症和衰老細胞負擔- 與上面討論的 NAD+ 前體可以增加 SASP 表達的發現相反。因此,NAD+對衰老的調節似乎很複雜。
總體而言,這些發現表明,在旨在恢復衰老過程中 NAD+ 水平的方法中,應考慮靶向免疫細胞,如 T 細胞、巨噬細胞和衰老細胞。然而,在更多地了解提高 NAD+ 水平的長期副作用之前,這項工作應該謹慎進行。
4.4. 神經變性
衰老與大多數神經退行性疾病密切相關,並伴隨著哺乳動物大腦中細胞 NAD+ 水平的降低。 NAD+ 耗竭在幾種表現出神經退行性變的加速衰老模型以及神經退行性疾病(包括阿爾茨海默病、帕金森病和肌萎縮側索硬化症 (ALS))中均有報導。在與年齡相關的神經退行性疾病期間,大腦中 NAD+ 丟失的根本原因和機制在很大程度上仍是未知的。然而,多條證據支持 NAD+ 的神經保護作用(圖 3Ab)。
首先,軸突變性是許多與年齡相關的神經元疾病的前兆,其特點是 NAD+ 快速消耗。在正常生理條件下,NAD+ 生物合成酶煙酰胺單核苷酸腺苷酸轉移酶 (NMNAT2)(圖 1;框 1)是軸突中的一種存活因子,由於其快速周轉,需要通過順行軸突運輸不斷補充。然而,在軸突變性過程中,NMNAT2 軸突運輸被阻斷,蛋白質的軸突池迅速降解,導致軸突中 NAD+ 的嚴重消耗。此外,NAD+ 消耗酶 SARM1 被軸突損傷激活,並通過促進 NAD+ 降解來介導軸突變性。這最初在沃勒變性緩慢 (Wlds) 小鼠中得到證實,這些小鼠受到軸突變性的保護,顯示缺乏 SARM1 表達和更高的神經元 NAD+ 水平,這是由於 NMNAT1 的嵌合融合蛋白的過表達,這允許其從細胞核中重新分佈到軸突,在那裡它可以替代 NMNAT2的活性。 SARM1 在促進軸突損傷中的作用已在其他幾個體內模型中得到證實。 Sarm1 敲除小鼠免受軸突變性的影響,並且可以挽救由缺乏 NMNAT2 引起的嚴重軸突生長缺陷和圍產期死亡。最近,在神經元中過度表達 SARM1 顯性陰性版本的轉基因小鼠模型顯示出軸突退化的顯著延遲,並暗示基因治療或靶向 SARM1 的小分子治療神經病的有希望的效果。
總體而言,Wlds 小鼠研究在很大程度上顯示了生物合成酶 NMNAT1、NMNAT2 和 NMNAT3的神經保護功能及其在包括帕金森病在內的幾種神經退行性疾病中的保護作用。然而,機制仍不清楚。除了上面討論的 SARM1 依賴性 NAD+ 降解的調節外,NMNAT 對它們的底物和 NAD+ 前體 NMN 的降解似乎可以保護軸突免於退化。與 NAD+ 的神經保護作用相反,據報導,前體 NMN 具有促進 SARM1 激活和循環 ADP-核糖生成的神經毒性作用,導致軸突破壞。然而,NMN 積累導致軸突變性的證據需要得到驗證並且仍然存在爭議。儘管如此,這種可能性引發了關於通過補充 NAD+ 前體如 NMN 來增加 NAD+ 合成的治療潛力的問題。
支持 NAD+ 神經保護作用的其他證據還包括使用 P7C3 的研究,P7C3 是一種氨丙基咔唑,據報導是 NAM 補救途徑中 NAMPT 的變構激活劑(BOX 1)。 P7C3 在帕金森病、阿爾茨海默病和 ALS158 的小鼠模型中被證明具有神經保護作用。此外,據報導,除 SARM1 之外的 NAD+ 消耗酶在與衰老相關的神經退行性疾病期間在細胞內 NAD+ 消耗中發揮作用。例如,CD38 表達在阿爾茨海默病進展過程中增加,缺乏 CD38 的阿爾茨海默病小鼠模型(Cd38 基因敲除小鼠)導致其大腦中 NAD+ 水平升高,顯示出較溫和的疾病表型。與 NAD+ 的神經保護作用一致,Cd38 基因敲除小鼠也可免受缺血性腦損傷引起的神經元細胞死亡。大腦中的多個細胞,包括小膠質細胞、星形膠質細胞、神經元和內皮細胞,表達 CD38 (REFS108,160),用炎性細胞因子處理小膠質細胞和星形膠質細胞可誘導 CD38 表達。與這一發現一致,CD38 表達與神經炎症相關,小鼠大腦中的促炎巨噬細胞/小膠質細胞數量較多。據報導,CD38 及其同源物 CD157 也會影響社會行為,這證實了 NAD+ 對神經元功能的功能影響。儘管沒有直接證據表明 CD38 在神經退行性疾病中具有因果作用,但如上所述,CD38 正在成為一種參與炎症和衰老的關鍵酶,這與神經退行性疾病密切相關。最後,PARP1 激活還與阿爾茨海默病和帕金森病 發病機制相關。使用各種阿爾茨海默病和帕金森病模型的體內研究表明,PARP1 的缺失可以防止腦功能障礙和認知能力下降。然而,在神經退行性變期間 PARP1 激活對 NAD+ 耗竭的貢獻在很大程度上仍未得到探索。
從綜合研究結果來看,越來越多的證據表明 NAD+ 是維持健康神經系統的中樞代謝物,並且可以影響多種腦細胞類型的生物學,這表明抵消與衰老相關的 NAD+ 水平下降可能是一種可行的治療方法用於治療神經退行性疾病。發現用 NAD+ 補充劑恢復 NAD+ 水平以及兩種生物合成酶 NAMPT 和 NMNAT1 的過表達可防止軸突退化。此外,NAD+ 前體 NR 和 NMN 可改善阿爾茨海默病大鼠和小鼠模型的神經元細胞健康、記憶和認知功能,並在黑腹果蠅帕金森病模型和 ALS 小鼠模型中顯示出神經保護特性.重要的是,現在有幾項臨床試驗正在進行中,使用 NAD+ 前體,尤其是 NR,來治療神經系統疾病並促進健康老齡化(表 2)。這些試驗無疑將擴大我們對人類神經退行性過程中 NAD+ 代謝的理解。
5.NAD+ 水平下降的治療靶向
在過去的二十年裡,NAD+ 在健康老齡化和長壽中的重要性已得到認可。不同動物模型(如秀麗隱桿線蟲、黑腹果蠅、囓齒動物和人類原代細胞)的臨床前研究已確定 NAD+ 水平隨年齡而下降,下降幅度為 10% 至 65%,具體取決於不同的器官和年齡。 NAD+ 可以通過飲食和生活方式選擇來調節(圖 4)。它也可以在藥理學上進行調節,到目前為止,已經探索了三種主要方法來增加 NAD+ 水平:膳食補充 NAD+ 前體參與 NAD+ 的補救途徑; NAD+生物合成酶的調節,特別是那些調節從頭合成和補救途徑的限速步驟的酶(分別為α-氨基-β-羧基粘康酸ε-半醛脫羧酶(ACMSD)和NAMPT);以及抑制參與 NAD+ 降解的酶,例如 PARPs 和 CD38。 NAD+ 水平的增加已在多種人類疾病小鼠模型中顯示出功效(表 1),導致在衰老過程中對人體進行 NAD+ 增強劑的大量臨床試驗(表 2)。考慮到本綜述的範圍,我們不會詳細介紹與 NAD+ 相關的臨床試驗,我們建議讀者閱讀對該主題的更全面的綜述。
大多數囓齒動物的臨床前研究表明 NAD+ 增強療法具有強大的轉化潛力。對人體的研究還不太先進,迄今為止,評估 NAD+ 前體的藥代動力學和毒理學的臨床試驗主要證明 NMN 和 NR 給藥是安全的,並且可以有效地增加健康志願者的 NAD+ 水平(表 2)。值得注意的是,使用 NR 的 I 期臨床試驗比使用 NMN 的多,這些試驗提供了相互矛盾的結果。一項試驗表明,短期服用 NR 對健康的老年人有一些有益效果,另一項試驗表明,對 ALS 患者有積極的效果。然而,NR 對肥胖的老年男性幾乎沒有影響(另見代謝功能障礙部分和表 2 中的討論)。因此,需要進一步的人體臨床研究來確定適當的劑量、治療期和長期毒理學結果,同時考慮參與者的多樣性,以更好地解決 NAD+ 增強策略的轉化問題。
5.1膳食補充劑
已經廣泛研究了不同 NAD+ 前體對酵母和秀麗隱桿線蟲的壽命和健康壽命的影響。在野生型酵母和秀麗隱桿線蟲中,低(微摩爾)濃度的 NR 以一種依賴於沉默調節蛋白的方式延長壽命。此外,在向野生型秀麗隱桿線蟲施用 NAM 時觀察到壽命延長。然而,超生理劑量的 NAM(1-5 mM,與延長壽命的實驗中使用的 200 μM 相比 190)也與酵母和秀麗隱桿線蟲的壽命縮短有關 190-192。因此,目前尚不清楚 NAM 管理在老齡化背景下是有益還是有害。這種差異的一個潛在解釋是 NAM 直接和間接地影響 NAD+ 功能。除了作為補救途徑中的直接 NAD+ 前體外,NAM 還是 NAD+ 分解代謝的副產物,在高毫摩爾濃度下,它已被證明可作為 NAD+ 依賴性酶的反饋抑製劑,例如 PARPs193 和 sirtuins194體外。然而,在幾項動物研究中,關於高劑量 NAM 的抑製作用的結果尚無定論 195。 NAM 對 NAD+ 活性的抑製作用的另一個可能原因可能是 NAM 水平的增加導致 NAM 甲基化的成比例增加。這反過來會影響甲基的細胞可用性,這對 DNA 甲基化和基因表達調節很重要 (BOX 2)。高水平的甲基化 NAM 與 2 型糖尿病、帕金森病和心髒病的發病機制有關。因此,需要進一步的研究來闡明最安全的 NAM 劑量和治療期。
提高 NAD+ 的策略也被用於延長與年齡有關的疾病的壽命。使用 NR 和/或 NMN 補充 NAD+ 也可有效延緩早衰樣表型和延長 A 組色素性乾皮病相關的秀麗隱桿線蟲模型、共濟失調毛細血管擴張症和 Cockayne 綜合徵以及 Werner 綜合徵的蠕蟲模型(a早衰症)。 NMN 治療顯著增加了共濟失調毛細血管擴張症小鼠模型中的 NAD+ 水平和最長壽命。 NR 治療還通過調節肌營養不良小鼠模型中的線粒體代謝來改善肌肉乾細胞功能197,198。
迄今為止,很少有研究調查 NMN、NAM 和 NR 對延長野生型小鼠的健康和/或壽命的影響。在一項研究中,從 5 個月大開始長期(1 年)餵養 NMN 的小鼠可提高全身胰島素敏感性、能量代謝和身體活動,並改善血脂狀況,而沒有明顯的毒性作用。在另一項研究中,長期服用 NAM 可以預防與高脂肪飲食相關的疾病,促進健康,但對壽命沒有顯著影響。最後,在老年小鼠中使用 NR 後,其壽命略有增加(5%)。反映這些結果,大多數使用 NAD+ 前體的臨床前研究都側重於通過對抗以 NAD+ 水平下降為特徵的不同年齡相關疾病來延長健康壽命(表 1)。已經證明,NMN 可增強各種器官的線粒體功能,包括骨骼肌、腎臟、肝臟、心臟、眼睛、大腦 和心血管系統,並且可以減輕脈管系統中的氧化應激,包括改善神經血管耦合反應。大腦皮層老化。補充 NMN 的這些作用可能是由 sirtuins 介導的。例如,SIRT3 在心肌病小鼠模型中介導 NMN 誘導的心臟和心外代謝功能的改善。 NMN 補充劑還通過 SIRT1 依賴性機制增加老年小鼠的血流量和耐力,從而增加內皮細胞數量並改善內皮細胞功能。
最後,已經表明,NR 的還原形式 NRH 在增加細胞內 NAD+ 含量方面比 NR 更穩定和有效,這是通過新的代謝途徑發生的,其中腺苷激酶將 NRH 轉化為 NMNH,然後將其氧化為 NMN或進一步被 NMNAT 代謝產生 NADH,然後產生 NAD+。因此,未來的研究將需要比較使用 NRH 與更常用的 NAD+ 前體 NAM、NR 和 NMN 作為治療劑的療效,以及其潛在的副作用。
總之,上面討論的 NAD+ 前體的有希望的臨床前結果表明,NAD+ 增強可以促進哺乳動物的健康壽命甚至壽命。 NAD+ 前體的這些有益作用可能與人類有關,目前正在臨床試驗中進行測試(表 2)。
5.2 NAD+生物合成的調節
NAD+ 的補救和從頭合成途徑也是提高體內 NAD+ 水平的治療的潛在目標。具體而言,NAMPT 的激活劑(NAD+ 補救途徑中的限速酶)和 NMNATs 有助於 NAD+ 從頭和補救途徑(圖 1),已被建議作為提高組織 NAD+ 水平的可能治療干預措施。事實上,神經保護劑 P7C3 增強了 NAMPT 活性並提高了多柔比星處理的人類細胞中的 NAD+ 水平,這表明它可能是衰老和與年齡相關的疾病過程(例如神經退行性變)的功能性治療劑。然而,P7C3 在增加 NAMPT 活性方面的真正功效仍然存在爭議。最近,另一種小分子 SBI-797812 被提議作為一種有效的 NAMPT 活化劑,在納摩爾範圍內具有活性。該化合物在體外增加了 NAMPT 介導的 NMN 產生,重要的是,增加了細胞系和體內的 NAD+ 水平。儘管 SBI-797812 在體內僅提高肝臟 NAD+ 水平的作用有限,但這是一種提高細胞內 NAD+ 水平的有前途的藥理學方法。此外,TES-991 和 TES-102524 對 ACMSD 的藥理學抑制促進了從頭 NAD+ 合成和 SIRT1 活性,最終增強了小鼠肝臟、腎臟和大腦中的線粒體功能。儘管這些策略有望在治療上提高 NAD+ 水平,但仍然存在的挑戰之一是更精確地確定其中一些分子(如 P7C3)如何在分子水平上發揮作用,確定它們的靶標並探索潛在的脫靶效應。
除了調節 NAD+ 水平外,近年來,人們還關注主要通過施用氧化還原循環醌 β-lapachone 來調節 NAD+/NADH 氧化還原平衡,這是一種 NAD(P)H:醌的外源性共底物受體氧化還原酶 1 (NQO1),從 NADH 再生 NAD+。 NQO1 顯示通過熱量限制增加(儘管它不足以介導這種飲食干預的抗衰老作用)217,並且通過施用 β-lapachone 增強 NQO1 活性可防止老年小鼠運動和認知功能的年齡依賴性下降通過改善線粒體功能障礙218,219。然而,NQO1 通常在大多數實體瘤中過度表達,其中 β-拉帕酮給藥會導致氧化還原循環和氧化應激失衡;因此,β-拉帕酮可能會根據具體情況產生多種影響,應謹慎使用。
5.3抑制 NAD+ 消耗
針對 NAD+ 降解酶和途徑可能是最近治療發展中最引人注目的領域。具體而言,靶向 PARPs 和 NADases,包括 CD38、CD157 和 SARMs,在治療與 NAD+ 水平下降相關的年齡相關疾病方面具有巨大潛力。在秀麗隱桿線蟲中,PARP 抑制導致野生型蠕蟲、共濟失調毛細血管擴張模型和高血糖條件下的壽命顯著延長。
PARP1 抑製劑,例如 olaparib 和 rucaparib,作為化療或癌症單一療法的輔助藥物上市銷售。它們使腫瘤對 DNA 損傷敏感,但由於毒性,它們的用途有限。 CD38 是哺乳動物中主要的 NADase 之一,在與年齡相關的 NAD+ 水平下降中起關鍵作用(參見衰老中的 NAD+ 依賴性機制部分)。幾種 CD38 抑製劑已經存在或正在開發中,其中一些可增強體內 NAD+ 水平。例如,芹菜素是一種天然存在的類黃酮,可提高人體細胞和小鼠肝組織中的 NAD+ 水平,並改善肥胖小鼠模型中的葡萄糖和脂質穩態。最近,芹菜素已被證明可下調 CD38 表達,並增加糖尿病大鼠腎臟中細胞內 NAD+/NADH 比率和 SIRT3 介導的線粒體抗氧化酶活性。另一種類黃酮 CD38 抑製劑木犀草素可在小鼠心肌缺血後提高 NAD+ 水平並保護內皮和心肌。臨床上,木犀草素對自閉症兒童具有神經保護作用,儘管在這種情況下並未直接評估 NAD+ 水平。此外,4-氨基喹啉的幾種衍生物,包括化合物 78c,可抑制 CD38 並提高小鼠肌肉、肝臟和心臟中的 NAD+ 水平230。化合物 78c 還可以防止小鼠中與年齡相關的 NAD+ 水平下降,用這種化合物治療老年小鼠可以改善代謝功能障礙、減少 DNA 損傷積累和改善肌肉功能。儘管研究作者沒有直接報告壽命變化,但他們確實報告了長壽 AMPK 通路的激活和 mTOR-p70S6K 和 ERK 通路的激活減少。最近,化合物 78c 保護小鼠免受缺血後內皮細胞和心肌細胞損傷。總體而言,越來越多的證據表明,靶向 CD38 和相關的 NAD+ 消耗酶,如 PARP,具有作為 NAD+ 促進靶標以延長人類健康壽命的巨大潛力,並且正在開發幾種藥理學方法來抑制 CD38 及其 NADase 活性 232(表 1 )。
6. 結論和觀點
在最初發現近 90 年後,NAD+ 正在成為衰老領域的中心代謝物,NAD+ 水平下降正在成為幾種與年齡相關的疾病的既定特徵。 NAD+ 領域發展迅速,並已發展成為生物醫學研究中令人興奮的主要研究領域之一。在過去的 5 年中,取得了許多進展,包括開發令人興奮的工具和技術(補充框 4),以進一步加深我們對 NAD+ 水平如何影響或受複雜信號傳導、代謝和細胞途徑影響的理解。此外,通過使用穩定同位素示踪和 NAD+ 生物傳感器,我們對 NAD+ 水平如何在細胞水平和系統水平進行調節的理解也取得了重大進展和改進。我們現在對導致 NAD+ 水平隨年齡下降的機制以及 NAD+ 消耗酶(如 CD38 和 SARM1)以及 PARP 在此過程中的新興作用有了更深入的了解。此外,現在已知這種 NAD+ 水平的下降會影響多種年齡依賴性細胞過程,包括 DNA 修復、氧化應激和免疫細胞功能。此外,最近的臨床前試驗表明,使用不同的動物模型,NAD+ 消耗是與年齡相關的疾病的關鍵途徑,包括神經退行性、代謝和早衰症。然而,目前尚不清楚哪些細胞和酶會導致特定疾病中 NAD+ 水平的下降。此外,可以利用哪些目標或途徑來有效和安全地恢復 NAD+ 穩態仍在研究中。好消息是,不同的 NAD+ 增強策略已被證明可有效延長健康和壽命(參見 NAD+ 水平下降的治療靶向部分和表 1)。因此,使用 NAD+ 前體(如 NMN 和 NR)以及促進 NAD+ 生物合成或抑制 NAD+ 降解的小分子,為治療與衰老相關的疾病和延長人類健康壽命提供了一種令人興奮的治療方法。這一點尤其重要,因為老年人口正在迅速增加,預計與老齡化相關的疾病將在未來幾十年造成巨大的社會和經濟負擔。然而,NAD+ 增強療法對人類的可轉化性仍然是有待回答的關鍵問題。正在進行幾項人體臨床試驗,以評估 NAD+ 增強的安全性和有效性(表 2),重要的是,短期 NR/NMN 給藥的早期試驗已證明它是安全的,並且可以增加健康參與者的 NAD+ 水平。然而,儘管初步結果令人鼓舞,但長期補充 NAD+ 前體是否有任何副作用仍然未知。此外,還需要回答許多其他問題,以加深我們對 NAD+ 增強療法潛力的理解:NMN 和 NR 是否有任何組織/疾病特異性?不同疾病所需的 NAD+ 前體治療劑量是多少?是否可以考慮將 NAD+ 增強策略(例如 CD38 和/或 PARP1 抑製劑與 NAD+ 前體補充劑)相結合?希望當前臨床試驗的即將到來的結果將闡明我們未解決的問題,並為破譯 NAD+ 在人類衰老過程中的作用的未來方向奠定基礎。.
肥胖是一種日益嚴重的全球流行病。肥胖者更容易患上以肥胖增加、胰島素抵抗、高血糖、高血壓和血脂異常為特徵的代謝疾病。因此,這些人患 2 型糖尿病、心血管疾病、非酒精性脂肪肝、動脈粥樣硬化、中風和癌症的風險更高。因此,肥胖會加速和加劇衰老,並與壽命縮短有關。在過去的二十年中,越來越多的證據表明,靶向 NAD+ 代謝可能會提供治療益處並有助於治療代謝疾病和衰老。
NAD+ 是通過調節酵母提取物的代謝率首次發現的,因此 NAD+ 與代謝之間的聯繫已為人所知近一個世紀。我們現在知道 NAD+ 位於新陳代謝的核心並調節多種代謝途徑的代謝通量(詳情參見框 1 和補充框 2)。因此,NAD+ 穩態是各種代謝組織正常功能所必需的,包括脂肪、肌肉、腸、腎和肝。然而,最近在釀酒酵母中發現了另一個關鍵發現,表明長壽蛋白 Sir2(酵母 sirtuin)以 NAD+ 依賴性方式表現出其延長壽命的作用。這一突破性發現表明 Sir2 的活性可能與代謝狀態有關。為了支持這一假設,現在已經在哺乳動物系統中證明了延長壽命的代謝操作,例如運動、熱量限制限時餵養和生酮飲食,以及健康的晝夜節律(包括定期睡眠模式 80),部分通過增加 NAD+ 水平起作用,這會導致去乙酰化酶的激活。
改變或破壞代謝狀態,例如高脂飲食、產後體重減輕和晝夜節律紊亂的結果,可導致 NAD+ 水平降低,從而降低去乙酰化酶和其他 NAD+ 的活性依賴的細胞過程(詳見補充框 2)。相反,最近顯示增加細胞 NAD+ 水平可以減少還原性壓力並推動代謝反應的方向性。此外,較高的 NAD+ 水平可以促進核 SIRT1 和線粒體 SIRT3 的去乙酰化酶和去乙酰化酶活性,從而調節線粒體功能並防止高脂飲食引起的代謝疾病。
總之,這些關鍵研究提供了強有力的證據和基本原理,即靶向 NAD+ 降解途徑或提高 NAD+ 水平可以影響代謝過程並可以預防代謝疾病。為了進一步支持該模型,許多研究現在表明,包括 Parp1 敲除和 Cd38 敲除小鼠在內的小鼠模型,或用 PARP 或 CD38 抑製劑治療的小鼠,具有超生理水平的 NAD+。它們還可以防止肥胖,提高代謝率,並且在高脂肪飲食條件和衰老過程中(相對)正常的葡萄糖代謝。此外,接受高脂肪飲食的小鼠炎症增加,導致 NAMPT 表達降低,NAD+ 補救途徑的活性降低 82 (BOX 1),為肥胖期間 NAD+ 水平下降的原因提供了潛在的機制解釋。根據這一機制,脂肪細胞特異性缺失 NAMPT 的小鼠其脂肪組織中的 NAD+ 水平較低,胰島素抵抗增加和代謝功能障礙增加,這可以通過補充 NMN 來挽救。此外,最近的多項研究表明,可以恢復與衰老相關的低 NAD+ 水平的 NAD+ 補充劑(NR 和 NMN)在囓齒動物模型中也可以預防肥胖,這表明這些補充劑可以作為治療方法來恢復人類肥胖患者的代謝健康。最近的幾項臨床試驗已經開始研究 NAD+ 前體在人類肥胖患者中改善代謝健康和葡萄糖代謝的功效。到目前為止,大多數這些研究都是在健康個體中完成的(見表 1),以測試可以安全提高 NAD+ 水平的 NR 和 NMN 的有效劑量。然而,最近的兩項隨機雙盲研究使用 NR 治療超重和肥胖患者 6 周和 12 週 。不幸的是,雖然 NR 可以有效增加這些人的 NAD+ 水平,但兩項研究的參與者都沒有表現出任何體重減輕、胰島素敏感性增加或線粒體功能增強等代謝參數的跡象。因此,目前尚不清楚靶向 NAD+ 代謝是否能有效治療肥胖或老年人的代謝疾病。
4.2.免疫細胞功能失調
炎症現在被認為是衰老的標誌和疾病的關鍵驅動因素(圖 3Aa),包括與衰老相關的疾病,並被描述為發病和死亡的主要風險因素。慢性炎症通過免疫細胞和代謝細胞(例如肝細胞和脂肪細胞)之間的複雜串擾,影響葡萄糖和脂質攝取以及胰島素敏感性等代謝過程,對全身代謝產生深遠影響。儘管在過去十年中人們對免疫代謝的興趣增加了,但人們對 NAD+ 如何影響慢性炎症和免疫細胞功能知之甚少。接下來我們將討論正在出現的畫面。
先天免疫。
慢性低度炎症,特徵為先天免疫系統異常激活,促炎細胞因子(如腫瘤壞死因子 (TNF)、IL-6 和 IL-1β)表達增強,以及免疫複合物(如 NLRP3 炎症小體)激活,現在被認為是衰老相關疾病和代謝疾病的關鍵驅動因素。此外,現在發現改變的巨噬細胞活化和表型極化是這種炎症的關鍵來源。例如,在內臟脂肪等肥胖組織中,外周單核細胞被受壓的脂肪細胞募集,導致促炎性 M1 樣巨噬細胞的進行性浸潤和駐留的抗炎 M2 巨噬細胞的移位。內臟脂肪中巨噬細胞極化狀態的這種轉變伴隨著促炎細胞因子的表達增強、胰島素抵抗和脂肪分解率的降低。 100 多年前,巨噬細胞的發現者 Elie Metchnikoff 是第一個觀察到衰老組織中巨噬細胞豐度增加的人。儘管這一觀察最初被忽視,但現在越來越多的證據表明,衰老不僅會導致巨噬細胞豐度增加,而且還會伴隨巨噬細胞極化狀態和功能的改變,這正在成為炎症的關鍵驅動因素。
一些表明 NAD+ 影響巨噬細胞功能的最早研究表明,抑制 NAMPT 和隨後消耗巨噬細胞中的 NAD+ 池會降低促炎細胞因子(例如 TNF)的分泌,並導致形態學變化,例如減少這些細胞中的擴散。最近的研究表明,NAD+ 是巨噬細胞功能的關鍵調節因子,巨噬細胞活化與 NAD+ 生物合成或降解途徑的上調有關,這取決於獲得的命運。例如,我們的實驗室最近表明,促炎 (M1) 巨噬細胞極化與 CD38 表達增強有關,從而導致 NAD+ 消耗增加。相反,抗炎 (M2) 巨噬細胞極化與依賴於 NAMPT45 的 NAD+ 水平的增加有關。阻斷 M1 巨噬細胞和 M2 巨噬細胞中的 NAM 補救途徑 (BOX 1) 顯著降低了與 M1 和 M2 表型相關的選定基因的基因表達。這種效果可以通過補充 NAD+ 前體 NMN 和 NR 來挽救,它們繞過和挽救 NAMPT 抑制。與 M1 巨噬細胞相比,M2 巨噬細胞需要更多的 NR/NMN 來拯救巨噬細胞活化,這表明 NAD+ 是一般巨噬細胞活化的關鍵代謝物,其代謝受到差異調節以控制 M1 和 M2 巨噬細胞中不同的生物過程和功能。
與我們的結果一致,最近的一項研究發現,M1 巨噬細胞極化與 NAD+ 的降解增強有關,抑制 NAMPT 可阻斷 M1 巨噬細胞的糖酵解轉變,限制體外促炎反應並減少體內對膿毒症的全身炎症反應。這項研究和最近的另一份報告表明,這種 NAD+ 轉換,特別是在巨噬細胞 M1 極化的最初幾個小時內,取決於活性氧誘導的 DNA 損傷和 PARP1 的激活。然而,我們實驗室最近發表的結果沒有檢測到在 M1 巨噬細胞極化期間 DNA 損傷或 PARP1 激活的證據。相比之下,我們的結果表明 CD38 是 M1 巨噬細胞中主要的 NAD+ 消耗酶。這些結果與最近的工作一致,表明 M1 巨噬細胞免受活性氧誘導的 DNA 損傷,這是由於參與抗氧化防禦的基因轉錄增加,如 SOD2 (REFS102-104)。因此,這些相互矛盾的觀察強調需要更好地描述促炎和抗炎巨噬細胞極化過程中 NAD+ 的主要消費者,以確定觀察到的 NAD+ 水平下降是否取決於背景和時間,並確定 NAD+ 水平影響促炎的分子機制和抗炎基因表達和巨噬細胞功能。
在衰老過程中,NAD+ 水平下降與肝臟和脂肪中促炎性 M1 樣常駐巨噬細胞的積累增加有關,其特點是 CD38 表達增加和 NADase 活性升高。通過體外和體內方法,發現這些過表達 CD38 的 M1 樣巨噬細胞直接被衰老細胞分泌的炎性細胞因子激活(稍後將詳細討論)。此外,衰老巨噬細胞的特徵是 NAD+ 從頭合成受損,這本身可能會影響衰老過程中的巨噬細胞功能6。
關於炎症,上述研究表明促炎 M1 樣巨噬細胞(除了衰老細胞;見下文)可能是衰老組織中促炎細胞因子的主要來源。衰老與主要由髓系免疫細胞表達的 NLRP3 炎症小體的激活增加相關,以及隨後 IL-1β 的表達增加,IL-1β 是與促進衰老相關疾病有關的主要細胞因子。促炎細胞因子的表達增強可能會導致炎症的惡性循環,導致更大的炎症、增強的組織和 DNA 損傷、進一步激活 NAD+ 的主要消費者,如 CD38 和 PARPs,並加速與年齡相關的生理衰退。
因此,靶向巨噬細胞免疫代謝途徑,特別是那些調節 NAD+ 生物合成或降解途徑的途徑,可以作為激活或抑制巨噬細胞功能和調節巨噬細胞極化狀態的治療策略進行探索。這當然與調節炎症相關,但也可以用於緩解由慢性炎症驅動的疾病,例如神經退行性疾病和自身炎症性疾病,也可以作為癌症的治療策略,其中巨噬細胞極化可以是腫瘤促進 (M2) 或腫瘤抑制性(M1)
適應性免疫。
與先天免疫系統一樣,衰老的特徵是由於適應性免疫細胞的功能改變(稱為免疫衰老)而導致建立有效適應性免疫反應的能力降低。
衰老導致免疫細胞群失衡或傾斜,包括初始 T 細胞和 B 細胞水平降低、T 細胞抗原受體多樣性喪失和虛擬記憶 T 細胞水平增加。 NAD+ 和 NAD+ 消耗酶在 T 細胞生物學中的調節作用已得到證實;然而,它們對適應性免疫老化的貢獻在很大程度上是未知的。一方面,細胞外 NAD+ 被認為是一種危險信號,可導致特定 T 細胞亞群(例如調節性 T 細胞)中的細胞死亡。另一方面,NAD+ 似乎表現出免疫調節特性,例如影響 T 細胞極化 。然而,NAD+ 是否促進特定的 T 細胞表型以及用 NAD+ 前體操縱 NAD+ 代謝是否會導致類似的免疫調節特性仍然未知。
適應性免疫衰老的一個既定標誌是高細胞毒性 CD8+CD28-T 細胞的記憶群體的擴展,其特徵是效應分子(如粒酶 B115)的高分泌。該細胞群的特點是 SIRT1 和 FOXO1 水平降低,從而導致糖酵解能力和顆粒酶 B 產量增強。這些研究強調了通過操縱與年齡相關的適應性免疫功能障礙中的 NAD+ 相關途徑進行代謝重編程的潛力。通過 CD38 抑制上調 NAD+-SIRT1-FOXO1 軸可增加 T 輔助細胞 1 和 17 雜合細胞的效應功能,未來使用 CD38 抑製劑或 NAD+ 前體的研究可能顯示靶向 NAD+ 在衰老適應性免疫系統中的治療潛力。
適應性免疫衰老的另一個免疫學特徵是耗盡的 T 細胞數量增加,其特徵是抑制性受體分子(例如 PD1 和 TIM3)的表達、增殖能力降低和效應功能降低 118,119。 PD1 是免疫檢查點的一個組成部分,其阻斷通常用作抗癌策略,但也有人提出恢復老化 T 細胞的效應功能。關於 NAD+ 代謝,最近的一項研究表明,CD38 過表達與 PD1 阻斷抗性癌症中功能失調和耗竭的 CD8 T 群體相關 123,124,突出了將 CD38 抑制研究擴展到與年齡相關的耗竭 T 細胞的潛力。然而,儘管非常有趣,但這一假設仍有待深入探討,需要更多的研究來確定這種方法的臨床前療效。
因此,總體而言,需要做更多的工作來確定操縱 NAD+ 水平是否能有效逆轉適應性免疫系統中與衰老相關的免疫功能障礙,同樣重要的是,這種操縱是否安全。
4.3. 細胞衰老
在衰老過程中,暴露於代謝、基因毒性或致癌基因誘導的壓力的細胞經歷了本質上不可逆的細胞週期停滯,稱為細胞衰老。衰老細胞的一種主要表型以及它們如何促進疾病是炎症介質(主要是細胞因子和趨化因子)的表達增加,稱為衰老相關分泌表型 (SASP),它通過乾擾幹細胞導致組織穩態受損再生、組織和傷口修復和炎症(圖 3Aa)。隨著衰老細胞的數量隨著年齡的增長而逐漸增加,細胞衰老與幾種與年齡相關的疾病有關,用藥物抗衰老劑清除衰老細胞可能是治療幾種以前無法治癒的疾病的有效方法,包括阿爾茨海默病。此外,旨在提高衰老過程中細胞 NAD+ 水平的治療是延長健康壽命的有希望的目標,但 NAD+ 如何影響細胞衰老尚不清楚。最近表明,衰老細胞上調了 NAM 補救酶 NAMPT(BOX 1)的表達,並且衰老細胞的 SASP 取決於 NAD+ 水平。用 NMN 治療衰老細胞可以提高 SASP,導致慢性炎症增加,並可以促進炎症驅動癌症的發展。這些發現表明,服用 NAD+ 增強補充劑,如 NR 和 NMN,可能會以長期副作用為代價,如增強慢性炎症和癌症發展。因此,更好地了解提高 NAD+ 水平的益處和無人關注的副作用將是未來研究和正在進行的臨床試驗的一個重要重點領域。由於炎症是一個非常複雜和多用途的過程,需要更多的研究來更好地了解 NAD+ 水平如何影響不同的炎症狀態以及在什麼情況下,並確定 NAD+ 代謝如何從機制上影響炎症免疫和衰老細胞的生物學。
儘管有充分證據表明衰老組織中衰老細胞的積累以及這些組織中 NAD+ 水平的下降,但沒有研究將炎症性衰老細胞的積累與衰老過程中的 NAD+ 水平聯繫起來。最近,研究表明哺乳動物組織中的 CD38 水平隨著年齡的增長而增加,並且 CD38 被認為是導致衰老過程中 NAD+ 水平下降的主要 NAD+ 消耗酶。然而,驅動衰老組織中 CD38 表達增加的機制以及哪些細胞在這些組織中表達 CD38 尚不清楚。最近的觀察表明,先天免疫細胞,尤其是巨噬細胞,可能是響應 SASP 並伴有 NAD+ 降解的主要細胞群,從而導致整個生物體的 NAD+ 水平下降。我們發現,在來自衰老細胞的條件培養基中共培養或暴露於條件培養基中的巨噬細胞增強了消耗 NAD+ 的酶 CD38 的表達並增加了增殖。重要的是,另一組也獨立地表明,衰老細胞及其 SASP 激活 CD38 表達並促進巨噬細胞中 CD38 依賴性 NADase 活性。此外,我們在衰老小鼠模型和用誘導衰老的化學治療試劑阿黴素治療的小鼠中發現,衰老細胞在代謝組織(例如內臟脂肪和肝臟)中的積累可以直接激活組織駐留巨噬細胞中 CD38 的表達。為了進一步支持炎症、衰老細胞負荷和 NAD+ 之間的聯繫,最近在小鼠身上進行的一項研究還表明,線粒體功能失調的細胞會啟動促炎程序,並分泌促炎細胞因子。這與更大的衰老細胞負擔、增加的代謝和身體功能障礙以及過早衰老有關。在這種情況下,補充 NR 能夠部分挽救這種多病綜合徵,部分是通過減少炎症和衰老細胞負擔- 與上面討論的 NAD+ 前體可以增加 SASP 表達的發現相反。因此,NAD+對衰老的調節似乎很複雜。
總體而言,這些發現表明,在旨在恢復衰老過程中 NAD+ 水平的方法中,應考慮靶向免疫細胞,如 T 細胞、巨噬細胞和衰老細胞。然而,在更多地了解提高 NAD+ 水平的長期副作用之前,這項工作應該謹慎進行。
4.4. 神經變性
衰老與大多數神經退行性疾病密切相關,並伴隨著哺乳動物大腦中細胞 NAD+ 水平的降低。 NAD+ 耗竭在幾種表現出神經退行性變的加速衰老模型以及神經退行性疾病(包括阿爾茨海默病、帕金森病和肌萎縮側索硬化症 (ALS))中均有報導。在與年齡相關的神經退行性疾病期間,大腦中 NAD+ 丟失的根本原因和機制在很大程度上仍是未知的。然而,多條證據支持 NAD+ 的神經保護作用(圖 3Ab)。
首先,軸突變性是許多與年齡相關的神經元疾病的前兆,其特點是 NAD+ 快速消耗。在正常生理條件下,NAD+ 生物合成酶煙酰胺單核苷酸腺苷酸轉移酶 (NMNAT2)(圖 1;框 1)是軸突中的一種存活因子,由於其快速周轉,需要通過順行軸突運輸不斷補充。然而,在軸突變性過程中,NMNAT2 軸突運輸被阻斷,蛋白質的軸突池迅速降解,導致軸突中 NAD+ 的嚴重消耗。此外,NAD+ 消耗酶 SARM1 被軸突損傷激活,並通過促進 NAD+ 降解來介導軸突變性。這最初在沃勒變性緩慢 (Wlds) 小鼠中得到證實,這些小鼠受到軸突變性的保護,顯示缺乏 SARM1 表達和更高的神經元 NAD+ 水平,這是由於 NMNAT1 的嵌合融合蛋白的過表達,這允許其從細胞核中重新分佈到軸突,在那裡它可以替代 NMNAT2的活性。 SARM1 在促進軸突損傷中的作用已在其他幾個體內模型中得到證實。 Sarm1 敲除小鼠免受軸突變性的影響,並且可以挽救由缺乏 NMNAT2 引起的嚴重軸突生長缺陷和圍產期死亡。最近,在神經元中過度表達 SARM1 顯性陰性版本的轉基因小鼠模型顯示出軸突退化的顯著延遲,並暗示基因治療或靶向 SARM1 的小分子治療神經病的有希望的效果。
總體而言,Wlds 小鼠研究在很大程度上顯示了生物合成酶 NMNAT1、NMNAT2 和 NMNAT3的神經保護功能及其在包括帕金森病在內的幾種神經退行性疾病中的保護作用。然而,機制仍不清楚。除了上面討論的 SARM1 依賴性 NAD+ 降解的調節外,NMNAT 對它們的底物和 NAD+ 前體 NMN 的降解似乎可以保護軸突免於退化。與 NAD+ 的神經保護作用相反,據報導,前體 NMN 具有促進 SARM1 激活和循環 ADP-核糖生成的神經毒性作用,導致軸突破壞。然而,NMN 積累導致軸突變性的證據需要得到驗證並且仍然存在爭議。儘管如此,這種可能性引發了關於通過補充 NAD+ 前體如 NMN 來增加 NAD+ 合成的治療潛力的問題。
支持 NAD+ 神經保護作用的其他證據還包括使用 P7C3 的研究,P7C3 是一種氨丙基咔唑,據報導是 NAM 補救途徑中 NAMPT 的變構激活劑(BOX 1)。 P7C3 在帕金森病、阿爾茨海默病和 ALS158 的小鼠模型中被證明具有神經保護作用。此外,據報導,除 SARM1 之外的 NAD+ 消耗酶在與衰老相關的神經退行性疾病期間在細胞內 NAD+ 消耗中發揮作用。例如,CD38 表達在阿爾茨海默病進展過程中增加,缺乏 CD38 的阿爾茨海默病小鼠模型(Cd38 基因敲除小鼠)導致其大腦中 NAD+ 水平升高,顯示出較溫和的疾病表型。與 NAD+ 的神經保護作用一致,Cd38 基因敲除小鼠也可免受缺血性腦損傷引起的神經元細胞死亡。大腦中的多個細胞,包括小膠質細胞、星形膠質細胞、神經元和內皮細胞,表達 CD38 (REFS108,160),用炎性細胞因子處理小膠質細胞和星形膠質細胞可誘導 CD38 表達。與這一發現一致,CD38 表達與神經炎症相關,小鼠大腦中的促炎巨噬細胞/小膠質細胞數量較多。據報導,CD38 及其同源物 CD157 也會影響社會行為,這證實了 NAD+ 對神經元功能的功能影響。儘管沒有直接證據表明 CD38 在神經退行性疾病中具有因果作用,但如上所述,CD38 正在成為一種參與炎症和衰老的關鍵酶,這與神經退行性疾病密切相關。最後,PARP1 激活還與阿爾茨海默病和帕金森病 發病機制相關。使用各種阿爾茨海默病和帕金森病模型的體內研究表明,PARP1 的缺失可以防止腦功能障礙和認知能力下降。然而,在神經退行性變期間 PARP1 激活對 NAD+ 耗竭的貢獻在很大程度上仍未得到探索。
從綜合研究結果來看,越來越多的證據表明 NAD+ 是維持健康神經系統的中樞代謝物,並且可以影響多種腦細胞類型的生物學,這表明抵消與衰老相關的 NAD+ 水平下降可能是一種可行的治療方法用於治療神經退行性疾病。發現用 NAD+ 補充劑恢復 NAD+ 水平以及兩種生物合成酶 NAMPT 和 NMNAT1 的過表達可防止軸突退化。此外,NAD+ 前體 NR 和 NMN 可改善阿爾茨海默病大鼠和小鼠模型的神經元細胞健康、記憶和認知功能,並在黑腹果蠅帕金森病模型和 ALS 小鼠模型中顯示出神經保護特性.重要的是,現在有幾項臨床試驗正在進行中,使用 NAD+ 前體,尤其是 NR,來治療神經系統疾病並促進健康老齡化(表 2)。這些試驗無疑將擴大我們對人類神經退行性過程中 NAD+ 代謝的理解。
5.NAD+ 水平下降的治療靶向
在過去的二十年裡,NAD+ 在健康老齡化和長壽中的重要性已得到認可。不同動物模型(如秀麗隱桿線蟲、黑腹果蠅、囓齒動物和人類原代細胞)的臨床前研究已確定 NAD+ 水平隨年齡而下降,下降幅度為 10% 至 65%,具體取決於不同的器官和年齡。 NAD+ 可以通過飲食和生活方式選擇來調節(圖 4)。它也可以在藥理學上進行調節,到目前為止,已經探索了三種主要方法來增加 NAD+ 水平:膳食補充 NAD+ 前體參與 NAD+ 的補救途徑; NAD+生物合成酶的調節,特別是那些調節從頭合成和補救途徑的限速步驟的酶(分別為α-氨基-β-羧基粘康酸ε-半醛脫羧酶(ACMSD)和NAMPT);以及抑制參與 NAD+ 降解的酶,例如 PARPs 和 CD38。 NAD+ 水平的增加已在多種人類疾病小鼠模型中顯示出功效(表 1),導致在衰老過程中對人體進行 NAD+ 增強劑的大量臨床試驗(表 2)。考慮到本綜述的範圍,我們不會詳細介紹與 NAD+ 相關的臨床試驗,我們建議讀者閱讀對該主題的更全面的綜述。
大多數囓齒動物的臨床前研究表明 NAD+ 增強療法具有強大的轉化潛力。對人體的研究還不太先進,迄今為止,評估 NAD+ 前體的藥代動力學和毒理學的臨床試驗主要證明 NMN 和 NR 給藥是安全的,並且可以有效地增加健康志願者的 NAD+ 水平(表 2)。值得注意的是,使用 NR 的 I 期臨床試驗比使用 NMN 的多,這些試驗提供了相互矛盾的結果。一項試驗表明,短期服用 NR 對健康的老年人有一些有益效果,另一項試驗表明,對 ALS 患者有積極的效果。然而,NR 對肥胖的老年男性幾乎沒有影響(另見代謝功能障礙部分和表 2 中的討論)。因此,需要進一步的人體臨床研究來確定適當的劑量、治療期和長期毒理學結果,同時考慮參與者的多樣性,以更好地解決 NAD+ 增強策略的轉化問題。
5.1膳食補充劑
已經廣泛研究了不同 NAD+ 前體對酵母和秀麗隱桿線蟲的壽命和健康壽命的影響。在野生型酵母和秀麗隱桿線蟲中,低(微摩爾)濃度的 NR 以一種依賴於沉默調節蛋白的方式延長壽命。此外,在向野生型秀麗隱桿線蟲施用 NAM 時觀察到壽命延長。然而,超生理劑量的 NAM(1-5 mM,與延長壽命的實驗中使用的 200 μM 相比 190)也與酵母和秀麗隱桿線蟲的壽命縮短有關 190-192。因此,目前尚不清楚 NAM 管理在老齡化背景下是有益還是有害。這種差異的一個潛在解釋是 NAM 直接和間接地影響 NAD+ 功能。除了作為補救途徑中的直接 NAD+ 前體外,NAM 還是 NAD+ 分解代謝的副產物,在高毫摩爾濃度下,它已被證明可作為 NAD+ 依賴性酶的反饋抑製劑,例如 PARPs193 和 sirtuins194體外。然而,在幾項動物研究中,關於高劑量 NAM 的抑製作用的結果尚無定論 195。 NAM 對 NAD+ 活性的抑製作用的另一個可能原因可能是 NAM 水平的增加導致 NAM 甲基化的成比例增加。這反過來會影響甲基的細胞可用性,這對 DNA 甲基化和基因表達調節很重要 (BOX 2)。高水平的甲基化 NAM 與 2 型糖尿病、帕金森病和心髒病的發病機制有關。因此,需要進一步的研究來闡明最安全的 NAM 劑量和治療期。
提高 NAD+ 的策略也被用於延長與年齡有關的疾病的壽命。使用 NR 和/或 NMN 補充 NAD+ 也可有效延緩早衰樣表型和延長 A 組色素性乾皮病相關的秀麗隱桿線蟲模型、共濟失調毛細血管擴張症和 Cockayne 綜合徵以及 Werner 綜合徵的蠕蟲模型(a早衰症)。 NMN 治療顯著增加了共濟失調毛細血管擴張症小鼠模型中的 NAD+ 水平和最長壽命。 NR 治療還通過調節肌營養不良小鼠模型中的線粒體代謝來改善肌肉乾細胞功能197,198。
迄今為止,很少有研究調查 NMN、NAM 和 NR 對延長野生型小鼠的健康和/或壽命的影響。在一項研究中,從 5 個月大開始長期(1 年)餵養 NMN 的小鼠可提高全身胰島素敏感性、能量代謝和身體活動,並改善血脂狀況,而沒有明顯的毒性作用。在另一項研究中,長期服用 NAM 可以預防與高脂肪飲食相關的疾病,促進健康,但對壽命沒有顯著影響。最後,在老年小鼠中使用 NR 後,其壽命略有增加(5%)。反映這些結果,大多數使用 NAD+ 前體的臨床前研究都側重於通過對抗以 NAD+ 水平下降為特徵的不同年齡相關疾病來延長健康壽命(表 1)。已經證明,NMN 可增強各種器官的線粒體功能,包括骨骼肌、腎臟、肝臟、心臟、眼睛、大腦 和心血管系統,並且可以減輕脈管系統中的氧化應激,包括改善神經血管耦合反應。大腦皮層老化。補充 NMN 的這些作用可能是由 sirtuins 介導的。例如,SIRT3 在心肌病小鼠模型中介導 NMN 誘導的心臟和心外代謝功能的改善。 NMN 補充劑還通過 SIRT1 依賴性機制增加老年小鼠的血流量和耐力,從而增加內皮細胞數量並改善內皮細胞功能。
最後,已經表明,NR 的還原形式 NRH 在增加細胞內 NAD+ 含量方面比 NR 更穩定和有效,這是通過新的代謝途徑發生的,其中腺苷激酶將 NRH 轉化為 NMNH,然後將其氧化為 NMN或進一步被 NMNAT 代謝產生 NADH,然後產生 NAD+。因此,未來的研究將需要比較使用 NRH 與更常用的 NAD+ 前體 NAM、NR 和 NMN 作為治療劑的療效,以及其潛在的副作用。
總之,上面討論的 NAD+ 前體的有希望的臨床前結果表明,NAD+ 增強可以促進哺乳動物的健康壽命甚至壽命。 NAD+ 前體的這些有益作用可能與人類有關,目前正在臨床試驗中進行測試(表 2)。
5.2 NAD+生物合成的調節
NAD+ 的補救和從頭合成途徑也是提高體內 NAD+ 水平的治療的潛在目標。具體而言,NAMPT 的激活劑(NAD+ 補救途徑中的限速酶)和 NMNATs 有助於 NAD+ 從頭和補救途徑(圖 1),已被建議作為提高組織 NAD+ 水平的可能治療干預措施。事實上,神經保護劑 P7C3 增強了 NAMPT 活性並提高了多柔比星處理的人類細胞中的 NAD+ 水平,這表明它可能是衰老和與年齡相關的疾病過程(例如神經退行性變)的功能性治療劑。然而,P7C3 在增加 NAMPT 活性方面的真正功效仍然存在爭議。最近,另一種小分子 SBI-797812 被提議作為一種有效的 NAMPT 活化劑,在納摩爾範圍內具有活性。該化合物在體外增加了 NAMPT 介導的 NMN 產生,重要的是,增加了細胞系和體內的 NAD+ 水平。儘管 SBI-797812 在體內僅提高肝臟 NAD+ 水平的作用有限,但這是一種提高細胞內 NAD+ 水平的有前途的藥理學方法。此外,TES-991 和 TES-102524 對 ACMSD 的藥理學抑制促進了從頭 NAD+ 合成和 SIRT1 活性,最終增強了小鼠肝臟、腎臟和大腦中的線粒體功能。儘管這些策略有望在治療上提高 NAD+ 水平,但仍然存在的挑戰之一是更精確地確定其中一些分子(如 P7C3)如何在分子水平上發揮作用,確定它們的靶標並探索潛在的脫靶效應。
除了調節 NAD+ 水平外,近年來,人們還關注主要通過施用氧化還原循環醌 β-lapachone 來調節 NAD+/NADH 氧化還原平衡,這是一種 NAD(P)H:醌的外源性共底物受體氧化還原酶 1 (NQO1),從 NADH 再生 NAD+。 NQO1 顯示通過熱量限制增加(儘管它不足以介導這種飲食干預的抗衰老作用)217,並且通過施用 β-lapachone 增強 NQO1 活性可防止老年小鼠運動和認知功能的年齡依賴性下降通過改善線粒體功能障礙218,219。然而,NQO1 通常在大多數實體瘤中過度表達,其中 β-拉帕酮給藥會導致氧化還原循環和氧化應激失衡;因此,β-拉帕酮可能會根據具體情況產生多種影響,應謹慎使用。
5.3抑制 NAD+ 消耗
針對 NAD+ 降解酶和途徑可能是最近治療發展中最引人注目的領域。具體而言,靶向 PARPs 和 NADases,包括 CD38、CD157 和 SARMs,在治療與 NAD+ 水平下降相關的年齡相關疾病方面具有巨大潛力。在秀麗隱桿線蟲中,PARP 抑制導致野生型蠕蟲、共濟失調毛細血管擴張模型和高血糖條件下的壽命顯著延長。
PARP1 抑製劑,例如 olaparib 和 rucaparib,作為化療或癌症單一療法的輔助藥物上市銷售。它們使腫瘤對 DNA 損傷敏感,但由於毒性,它們的用途有限。 CD38 是哺乳動物中主要的 NADase 之一,在與年齡相關的 NAD+ 水平下降中起關鍵作用(參見衰老中的 NAD+ 依賴性機制部分)。幾種 CD38 抑製劑已經存在或正在開發中,其中一些可增強體內 NAD+ 水平。例如,芹菜素是一種天然存在的類黃酮,可提高人體細胞和小鼠肝組織中的 NAD+ 水平,並改善肥胖小鼠模型中的葡萄糖和脂質穩態。最近,芹菜素已被證明可下調 CD38 表達,並增加糖尿病大鼠腎臟中細胞內 NAD+/NADH 比率和 SIRT3 介導的線粒體抗氧化酶活性。另一種類黃酮 CD38 抑製劑木犀草素可在小鼠心肌缺血後提高 NAD+ 水平並保護內皮和心肌。臨床上,木犀草素對自閉症兒童具有神經保護作用,儘管在這種情況下並未直接評估 NAD+ 水平。此外,4-氨基喹啉的幾種衍生物,包括化合物 78c,可抑制 CD38 並提高小鼠肌肉、肝臟和心臟中的 NAD+ 水平230。化合物 78c 還可以防止小鼠中與年齡相關的 NAD+ 水平下降,用這種化合物治療老年小鼠可以改善代謝功能障礙、減少 DNA 損傷積累和改善肌肉功能。儘管研究作者沒有直接報告壽命變化,但他們確實報告了長壽 AMPK 通路的激活和 mTOR-p70S6K 和 ERK 通路的激活減少。最近,化合物 78c 保護小鼠免受缺血後內皮細胞和心肌細胞損傷。總體而言,越來越多的證據表明,靶向 CD38 和相關的 NAD+ 消耗酶,如 PARP,具有作為 NAD+ 促進靶標以延長人類健康壽命的巨大潛力,並且正在開發幾種藥理學方法來抑制 CD38 及其 NADase 活性 232(表 1 )。
6. 結論和觀點
在最初發現近 90 年後,NAD+ 正在成為衰老領域的中心代謝物,NAD+ 水平下降正在成為幾種與年齡相關的疾病的既定特徵。 NAD+ 領域發展迅速,並已發展成為生物醫學研究中令人興奮的主要研究領域之一。在過去的 5 年中,取得了許多進展,包括開發令人興奮的工具和技術(補充框 4),以進一步加深我們對 NAD+ 水平如何影響或受複雜信號傳導、代謝和細胞途徑影響的理解。此外,通過使用穩定同位素示踪和 NAD+ 生物傳感器,我們對 NAD+ 水平如何在細胞水平和系統水平進行調節的理解也取得了重大進展和改進。我們現在對導致 NAD+ 水平隨年齡下降的機制以及 NAD+ 消耗酶(如 CD38 和 SARM1)以及 PARP 在此過程中的新興作用有了更深入的了解。此外,現在已知這種 NAD+ 水平的下降會影響多種年齡依賴性細胞過程,包括 DNA 修復、氧化應激和免疫細胞功能。此外,最近的臨床前試驗表明,使用不同的動物模型,NAD+ 消耗是與年齡相關的疾病的關鍵途徑,包括神經退行性、代謝和早衰症。然而,目前尚不清楚哪些細胞和酶會導致特定疾病中 NAD+ 水平的下降。此外,可以利用哪些目標或途徑來有效和安全地恢復 NAD+ 穩態仍在研究中。好消息是,不同的 NAD+ 增強策略已被證明可有效延長健康和壽命(參見 NAD+ 水平下降的治療靶向部分和表 1)。因此,使用 NAD+ 前體(如 NMN 和 NR)以及促進 NAD+ 生物合成或抑制 NAD+ 降解的小分子,為治療與衰老相關的疾病和延長人類健康壽命提供了一種令人興奮的治療方法。這一點尤其重要,因為老年人口正在迅速增加,預計與老齡化相關的疾病將在未來幾十年造成巨大的社會和經濟負擔。然而,NAD+ 增強療法對人類的可轉化性仍然是有待回答的關鍵問題。正在進行幾項人體臨床試驗,以評估 NAD+ 增強的安全性和有效性(表 2),重要的是,短期 NR/NMN 給藥的早期試驗已證明它是安全的,並且可以增加健康參與者的 NAD+ 水平。然而,儘管初步結果令人鼓舞,但長期補充 NAD+ 前體是否有任何副作用仍然未知。此外,還需要回答許多其他問題,以加深我們對 NAD+ 增強療法潛力的理解:NMN 和 NR 是否有任何組織/疾病特異性?不同疾病所需的 NAD+ 前體治療劑量是多少?是否可以考慮將 NAD+ 增強策略(例如 CD38 和/或 PARP1 抑製劑與 NAD+ 前體補充劑)相結合?希望當前臨床試驗的即將到來的結果將闡明我們未解決的問題,並為破譯 NAD+ 在人類衰老過程中的作用的未來方向奠定基礎。.
